{kind=link}
Immunogenicity of a trivalent haemorrhagic fever vaccine candidate against Sudan virus, Marburg virus and Lassa virus in an mpox vaccine
J Gen Virol. 2025 Oct;106(10). doi: 10.1099/jgv.0.002157.
ABSTRACT
A multivalent vaccine targeting high-consequence infectious diseases in Sub-Saharan Africa (SSA), which are linked to high mortality, morbidity and overlapping clinical manifestations, would significantly improve health security and economic stability in this region. Trivalent vector vaccines were devised to deliver digitally optimized versions of Orthoebolavirus, Orthomarburgvirus glycoproteins (GPs) and a Lassa mammarenavirus (LASV) nucleoprotein (NP) by a single Modified Vaccinia Ankara (MVA) known to protect against mpox virus (MPXV) along with a matched DNA vaccine. Three immunizations in mice and Hartley guinea pigs with MVA only or a DNA prime followed by two MVA administrations induced comparable levels of binding antibodies and LASV-specific T-cells, respectively. While DNA priming mitigated MVA-specific antibody responses, GP- and NP-specific antibodies developed already after a single MVA vaccination. Although a post-outbreak Ebola virus vaccine is available, outbreaks by other filoviruses, annual LASV epidemics and increased incidence of MPXV infections support the rationale for an MVA-based trivalent haemorrhagic fever vaccine for endemic and high-risk human populations in SSA.
PMID:41051941 | DOI:10.1099/jgv.0.002157
A Single Institution Retrospective Comparison of Two Radiotherapy Protocols for the Palliative Treatment of Canine Nasal Carcinoma
Vet Radiol Ultrasound. 2025 Nov;66(6):e70097. doi: 10.1111/vru.70097.
ABSTRACT
Optimal radiation protocols for canine nasal carcinoma are not established. Co-morbidities, access, and owner compliance can influence scheduling. Between 2015 and 2022, two radiotherapy protocols were used in the palliative treatment of canine nasal carcinoma at a single institution. Group A comprised 17 cases receiving 40 Gy in ten 4 Gy fractions delivered Monday, Wednesday, and Friday. Epistaxis was present in 11/17 (65%) cases. Median survival time (MST) was 298 days (95% CI: 163.54-432.45); progression-free survival was 173 days (95% CI: 117.87-228.12). Group B comprised 24 cases receiving 36 Gy in six 6 Gy fractions delivered Monday and Friday. Epistaxis was present in 20/24 (83%) cases. MST was 375 days (95% CI: 240.73-509.27); progression-free survival was 243 days (95% CI: 138.42-347.58). Dogs with Adams Stage 1 disease had the longest median overall (593 days) and progression-free survival (609 days). Four cases each received additional radiation treatment and/or toceranib at relapse. Palliative radiation therapy achieved control of clinical signs in the majority of cases, with an overall response rate of 100% (Group A) and 96% (Group B). In a multivariate Cox regression model with backwards elimination, when cases were stratified for tumor stage, neither the presence of epistaxis nor treatment (6 vs. 10 fractions) was independently associated with significant improvements in survival. Epistaxis at presentation did not appear to influence survival. These results indicate that palliative radiation therapy is highly effective in controlling clinical signs associated with nasal carcinoma. Increasing fractionation may have a limited effect on survival outcome or toxicity in the palliative setting.
PMID:41047698 | DOI:10.1111/vru.70097
Editorial: Strategies for mitigating zoonotic influenza outbreaks: a comprehensive preparedness approach
Front Public Health. 2025 Sep 3;13:1685224. doi: 10.3389/fpubh.2025.1685224. eCollection 2025.
NO ABSTRACT
PMID:40969630 | PMC:PMC12441160 | DOI:10.3389/fpubh.2025.1685224
Veterinary Nurse - Small Animal Wing (Part Time)
Salary £26,226 - £29,986 (pro rata for 0.96 FTE) + 15% Shift Allowance = £30,160 - £34,484 per annum. This equates to an hourly rate of £13.50-£15.52 plus a 15% uplift.
We have an exciting opportunity for a Registered Veterinary Nurse to join our nursing team at the Queens Veterinary School Hospital.
The referral hospital is a very fast-paced environment which requires you to be adaptable with the ability to work well under pressure. The role will be to provide nursing to an excellent standard in the Small Animal Wing. The small animal wing consists of four dog, two cat, critical care and isolation wards housing an average of 15-25 patients during the overnight period.
We are looking for nurses who can cover two 12 hour shifts (2100-0900hrs) every 7 weeks. Rotas are planned well in advance to help employees maintain a healthy work-life balance.
The role in Small Animal inpatients will include surgical, medical and critical care nursing (training can be given in critical care procedures). Cleaning, hygiene and administration duties linked to inpatient care will form a reasonable percentage of the time. This is a varied role where no two days are the same, so we are seeking individuals with a passion for nursing, with the ability to communicate with all levels of staff, students and clients. The ability to work on your own initiative with minimal supervision is essential. Shifts have Veterinary Intern and Veterinary Care Assistant support.
In return, we offer an encouraging and nurturing environment and have a dedicated team of clinicians and nurses who are committed to providing the best care for our patients.
Benefits:
- Generous paid annual leave including bank holidays
- Defined benefit pension scheme
- Enhanced family friendly policies
- Access to a dedicated Personal and Professional Development team
- Wellness programme including Occupational Health team and Staff counselling
- Staff discount scheme including shopping vouchers
- Cycle to work scheme
- Travel to work loans
- Eye care voucher scheme
- Discounted gym membership
- CPD allowance
If you have any questions about this role, please contact the Clinical HR Team on qvsh.hr@vet.cam.ac.uk. Please quote reference PP47333 on your application and in any correspondence about this vacancy.
Click the 'Apply' button below to register an account with our recruitment system (if you have not already) and apply online.
The closing date is midnight on Sunday, 12 October 2025.
In instructions for applicants: Applications will be monitored regularly, and we may contact candidates prior to the closing date. We reserve the right to close this vacancy early if we receive sufficient applications for the role. Therefore, if you are interested, please submit your application as early as possible.
Once an offer of employment has been accepted, the successful candidate will be required to undergo a health assessment.
The University actively supports equality, diversity and inclusion and encourages applications from all sections of society.
The University has a responsibility to ensure that all employees are eligible to live and work in the UK.
Antimicrobial use differs between general practice and referral settings in United Kingdom companion animals: a 12-month prospective study
Am J Vet Res. 2025 Sep 12:1-10. doi: 10.2460/ajvr.25.06.0229. Online ahead of print.
ABSTRACT
OBJECTIVE: To characterize the use of antimicrobials and frequency of microbial identification testing in cases seen by companion animal general practitioners and a single United Kingdom (UK) internal medicine referral service.
METHODS: This was a prospective, observational study of animals referred to a single UK-based internal medicine referral service over a 12-month period. Information recorded at the time of presentation included presenting complaint, current medications, and whether a microbial identification test had been performed. At discharge, the same information was recorded again alongside the final diagnosis.
RESULTS: 516 dogs and cats were enrolled, and 22.9% of cases were receiving antimicrobials at the time of presentation compared to 22.0% of cases at the time of discharge. Cases receiving antimicrobials at admittance were 2.7 (95% CI, 1.7 to 4.3) times more likely to be discharged on antimicrobials. Postreferral respiratory and urinary cases were 7.3 times more likely (OR, 7.3; 95% CI, 3.6 to 15.0) and 4.2 times more likely (OR, 4.2; 95% CI, 1.9 to 9.4), respectively, to be discharged on antimicrobials. Bacterial culture was more commonly performed within the referral center (72.1%) than prior to referral (16.9%).
CONCLUSIONS: Bacterial culture is performed more frequently by referral internal medicine clinicians than UK general practitioners. There are differences in the distribution of cases prescribed antimicrobials between these 2 groups of veterinarians.
CLINICAL RELEVANCE: This study provides information on antimicrobial prescribing patterns in companion animals across both general practice and referral settings in the UK. It highlights areas for antimicrobial stewardship improvement as well as identifies factors associated with antimicrobial prescription.
PMID:40939627 | DOI:10.2460/ajvr.25.06.0229
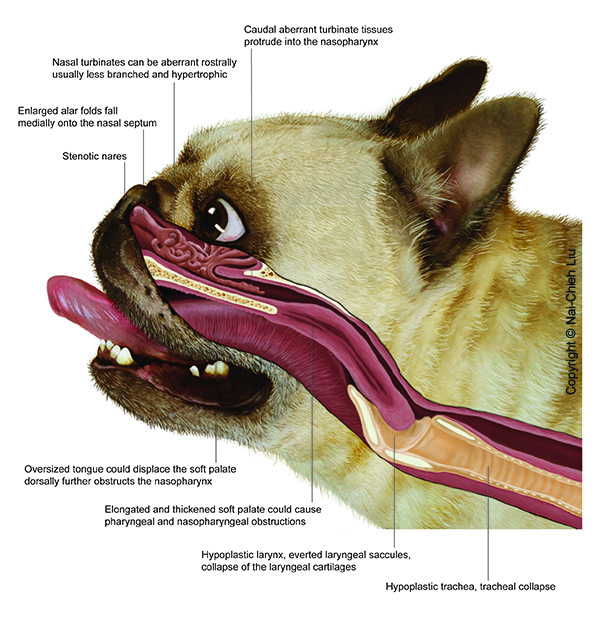
Pterygoid bone malformation and its limitations on the effectiveness of brachycephalic airway corrective surgery in brachycephalic dogs
J Small Anim Pract. 2025 Sep 11. doi: 10.1111/jsap.70028. Online ahead of print.
ABSTRACT
OBJECTIVES: This study aimed to examine the association between pterygoid bone medialisation and treatment outcomes after upper airway surgery in three brachycephalic breeds.
MATERIALS AND METHODS: Dogs that underwent CT of the head followed by routine surgery for brachycephalic obstructive airway syndrome were recruited in this study. Measurements obtained from the CT images included the width of the basisphenoid bone, interpterygoid distance and cross-sectional area of the nasopharynx. A ratio of width of the basisphenoid bone to interpterygoid distance allowed quantification of pterygoid bone medialisation. Pearson's correlations were calculated to assess the relationship between width of the basisphenoid bone: interpterygoid distance and cross-sectional area of the nasopharynx. Additionally, width of the basisphenoid bone: interpterygoid distance was compared across breeds, as well as between dogs with good and poor surgical outcomes (post-operative brachycephalic obstructive airway syndrome index ≥50%).
RESULTS: One hundred and forty-four brachycephalic dogs (47 Pugs, 64 French Bulldogs and 33 Bulldogs) and 30 non-brachycephalic controls were included in the analysis. The width of the basisphenoid bone: interpterygoid distance ratio in brachycephalic dogs was significantly higher (1.982 ± 0.379) than that of controls (1.646 ± 0.239, P < 0.001). A negative correlation was observed between width of the basisphenoid bone: interpterygoid distance and cross-sectional area of the nasopharynx in Pugs (ρ = -0.29, P = 0.048), French Bulldogs (ρ = -0.47, P < 0.001), Bulldogs (ρ = -0.71, P < 0.001) and controls (ρ = -0.55, P = 0.002). French Bulldogs with poor surgical outcomes exhibited a significantly higher width of the basisphenoid bone: interpterygoid distance (2.366 ± 0.327) than those with good surgical outcomes (1.813 ± 0.271, P < 0.0001).
CLINICAL SIGNIFICANCE: Pterygoid bone medialisation is associated with nasopharyngeal narrowing, which limits the effectiveness of surgical interventions in brachycephalic obstructive airway syndrome in affected French Bulldogs. As there are no surgical options currently reported to address this condition, these findings are important in guiding clinicians in providing prognostic information to owners during elective brachycephalic obstructive airway syndrome surgery.
PMID:40935636 | DOI:10.1111/jsap.70028
Junior Clinical Training Scholar in Small Animal Medicine
SCHOLARSHIP AWARD: £21,970.00 (TAX EXEMPT) per annum (Subject to change)
One-Year Junior Clinical Training Scholarship in Small Animal Medicine (Medicine Internship) Start date: 8 December 2025 (or as soon as possible thereafter)
We are pleased to offer a one-year Junior Clinical Training Scholarship in Small Animal Medicine, designed for veterinary graduates who wish to develop their knowledge and clinical skills in this specialty.
This intensive, hands-on programme provides broad exposure to all areas of small animal medicine, including medical oncology, medical neurology, diagnostic imaging, and clinical pathology. Structured practical and theoretical training is delivered under the supervision of experienced specialists.
As part of the role, you will contribute to the Department's clinical service, including participation in the out-of-hours rota, and support the small-group teaching of veterinary students. The programme offers extensive experience with a wide variety of medical cases and procedures, making it an excellent stepping stone towards a residency or advanced clinical practice.
What We Offer
- Competitive tax-free stipend including onsite accommodation (available for £300 per month including bills) in Central Cambridge
- CPD allowance and encouragement to attend and present at scientific meetings
- Good work-life balance with manageable out of hours duties on a shared rota
- 2 weeks of elective/dedicated research time on top of holidays
- Academic opportunities, e.g. teaching Cambridge students during rotations and College supervision opportunities; weekly department research and clinical seminars; journal and book clubs
- Proven track-record with publications and research projects with guidance on presentation and scientific writing skills.
- Assigned supervisor: - regular progress meetings, interview practice, provision of professional references and CV/cover letter proof reading by experienced senior clinicians to aid residency applications
- University library and extensive journal access
Who Can Apply
Applicants must be Members of the Royal College of Veterinary Surgeons or hold a veterinary degree that qualifies them for membership. A minimum of one years' experience in a UK-based primary care small animal practice, during which you have gained knowledge of UK veterinary regulations and practices, is essential. Applicants should have completed their VetGDP.
Interested?
We would welcome anyone wishing to apply for this scholarship to arrange a visit to the hospital to meet the team and find out more. To arrange a visit to the hospital and for informal enquiries about the scholarship programme. Please contact to Nick Bexfield, Clinical Director of Small Animal Services, by email: nb289@cam.ac.uk.
For general enquires please direct these to the HR Team via: vetmed@vet.cam.ac.uk.
How to Apply
Download the application form (JCTS1) and information pack from our website: www.vet.cam.ac.uk/job
Submit your completed application form (JCTS1), CV, and Covering Letter (outlining your motivation for applying) as one attachment via email to: vetmed@vet.cam.ac.uk
Deadline: Midnight on 02 October 2025
Important Information
Please note: The ability to take up this Scholarship is contingent upon you being able to evidence your right to work in the UK, or through gaining the right to work via the UK immigration system. Evidence will need to be provided before an offer can be made. Regrettably, this Scholarship is not suitable for sponsorship via the Skilled Worker or Temporary Worker visa routes as the minimum requirements cannot be met. For more information about the Department and the role please visit www.vet.cam.ac.uk.
Use of image-guided robotic-assisted drilling for transcondylar screw placement in the canine humerus
Vet Surg. 2025 Sep 5. doi: 10.1111/vsu.70010. Online ahead of print.
ABSTRACT
OBJECTIVE: To determine if a novel robotic system has comparable positional and angular accuracy to that achievable with patient-specific guides (PSG) when used for transcondylar screw (TCS) placement in the canine humerus.
STUDY DESIGN: Experimental laboratory study.
SAMPLE POPULATION: A total of 32 synthetic humeral models (16 per group).
METHODS: Bone models were three-dimensional (3D)-printed and drilled with the aid of a custom PSG or with the assistance of an image-guided surgical robot. A 2.5-mm hole was drilled medial to lateral and the entry point, exit point and angular trajectory of the drill hole were measured on postoperative computed tomography (CT) scans. Absolute differences between planned and actual positions and trajectories were compared between PSG and Robot groups.
RESULTS: None of the drill holes in this study violated the articular surface of the humerus. Entry point positioning was significantly more accurate in the PSG group, but drill hole trajectories (angulation) were more accurate in the Robot group. Exit point positioning was similar in the two groups.
CONCLUSION: Robotic assistance enables safe placement of drill holes for TCS. PSG enable more accurate drill entry, but robotic assistance allows for more accurate overall drill hole trajectory.
CLINICAL SIGNIFICANCE: Robotic assistance allows for accurate and safe drilling of screw holes for TCS placement in the humerus. The robotic procedure allows for a more limited surgical exposure, but the technical feasibility and outcomes associated with this approach should now be evaluated in cadavers before moving to clinical evaluation in live patients.
PMID:40913298 | DOI:10.1111/vsu.70010
Use of ultrasound to estimate tracheal diameter in unclipped brachycephalic dogs: A pilot study
Vet Rec Open. 2025 Sep 2;12(2):e70018. doi: 10.1002/vro2.70018. eCollection 2025 Dec.
ABSTRACT
BACKGROUND: A significant contributory factor to the brachycephalic obstructive airway syndrome index of bulldogs is the diameter of their tracheas. Bulldogs are predisposed to tracheal hypoplasia. A non-invasive, financially reasonable and widely accessible screening test for tracheal diameter would be useful for assessing the most appropriate dogs to breed from within bulldog populations and may help in predicting results following upper airway surgery.
METHOD: A prospective method comparison study involving 10 client-owned brachycephalic dogs was conducted. Each patient underwent tracheal ultrasonography using a handheld ultrasound device (Butterfly IQ+) followed by extubated computed tomography (CT) scanning of the head and neck. Ultrasonographic tracheal measurements were compared with CT tracheal measurements and analysed for agreement, accuracy, and inter-observer and intra-observer repeatability.
RESULTS: Bland‒Altman analysis showed there was good agreement between the ultrasound and CT tracheal measurements; however, the 95% limits of agreement were wide (+0.43 and ‒0.29 cm), indicating that the ultrasound measurements lacked a high level of accuracy. Ultrasound in general overestimated the tracheal diameter by 0.07 cm (p < 0.05). Intra-observer repeatability (mean range: 0.12 cm, average coefficient of variation [COV]: observer one;7.36%, observer two;5.53%, observer three;6.10%) was more consistent than inter-observer repeatability (mean range: 0.26 cm, average COV: 8.47%).
CONCLUSION: The accuracy of tracheal diameter measurements using an affordable handheld ultrasound device in unclipped brachycephalic patients was relatively low. However, technique modifications may significantly improve results, and further investigation is warranted to explore the utility of this method as a screening tool for tracheal hypoplasia in bulldog populations.
PMID:40904891 | PMC:PMC12403226 | DOI:10.1002/vro2.70018
Junior Clinical Training Scholar in Small Animal Studies
A scholarship is available to start on Monday 01 December 2025, or soon after, for a duration of 12.5 months.
SCHOLARSHIP AWARD: £21,970.00 (TAX EXEMPT) per annum.
University accommodation package, see details below.
Applications are invited for this one-year post-graduate training programme based in the Queen's Veterinary School Small Animal Hospital. On site accommodation is available for £300 per month including bills.
Junior Clinical Training Scholars will receive training and tuition as they rotate through anaesthesia, cardiology, diagnostic imaging, orthopaedics, dermatology, internal medicine, neurology, oncology, clinical pathology and soft tissue surgery and be supervised by recognised specialists in each field. Scholars will also have responsibility for primary care cases, and be involved in supervision and guidance of final year veterinary students. Scholars will be an integral part of the out of hours care of animals within the hospital, especially within the intensive care unit.
Summary of benefits
- High residency success rate - 74% of our interns have gone on to do a residency and 100% of interns who have been pursuing a residency have successfully achieved a residency or completed a specialist internship programme. We have 50 successful residency applications from intern cohorts 2015-2022 and 22 diplomates, so far!
- Competitive tax-free stipend including accommodation in Central Cambridge and bills included package
- Truly rotating internship through all specialties including flexibility to pursue extra time in rotations of your choice!
- Good work-life balance with manageable weekend and night work
- University library and journal access
- Monthly seminars with complimentary food and drink!
- Minimum of 2 weeks of elective/dedicated research time on top of holidays
- Academic opportunities, e.g. teach Cambridge students during rotations and College supervision opportunities; weekly department research and clinical seminars; journal and book clubs
- Proven track-record with publications and research projects with guidance on presentation and scientific writing skills.
- Assigned intern supervisor: - regular progress meetings, interview practice, provision of professional references and CV/cover letter proof reading by experienced senior clinicians to aid residency applications
- Generous CPD allowance and encouragement to present at scientific meetings
- RECOVER CPR training
- First opinion service including surgical cases
- A number of service-specific internships and residency opportunities encourage career progression following internship
Candidates must be Members of the Royal College of Veterinary Surgeons (RCVS) and the following skills and experience are desirable: - Surgical experience in dogs and cats - Completion of 1 year in primary care veterinary practice in the UK - For applicants for whom English is not their first language, a score of 7.5 in IELTS (with no element under 7), or a score of 100 in TOEFL (with no element less than 24).
For further benefits and details on the Internship: https://www.vet.cam.ac.uk/study/cts/jcts1/smallanimal
Informal enquiries should be directed to the Internship Directors, by email: internship.enquiries@vet.cam.ac.uk.
Please note:
The ability to take up this Scholarship is contingent upon you being able to evidence your right to work in the UK, or through gaining the right to work via the UK immigration system. Evidence will need to be provided before an offer can be made. Regrettably, this Scholarship is not suitable for sponsorship via the Skilled Worker or Temporary Worker visa routes as the minimum requirements cannot be met.
A JCTS Application Form (JCTS 1) and Information Pack can be downloaded from the following website: https://www.vet.cam.ac.uk/job
Applicants should supply a completed Junior Clinical Training Scholarship Application Form (JCTS 1), a CV and Covering Letter giving reasons for wishing to undertake the JCTS in the Department of Veterinary Medicine, University of Cambridge.
Applications should be submitted via e-mail to vetmed@vet.cam.ac.uk with the above documents as one attachment, by the closing date stated.
Please quote reference PP47176 on your application and in any correspondence about this vacancy.
The closing date for applications is midnight on 17 September 2025.
Applications will be monitored regularly, and we may contact candidates prior to the closing date. We reserve the right to close this vacancy early if we receive sufficient applications or extend the closing date if necessary. Therefore, if you are interested, please submit your application as early as possible.
The University actively supports equality, diversity and inclusion and encourages applications from all sections of society.
Modelling of strategies for the introduction and routine use of multivalent meningococcal conjugate vaccines (MMCVs) in the African meningitis belt
PLoS One. 2025 Aug 29;20(8):e0330627. doi: 10.1371/journal.pone.0330627. eCollection 2025.
ABSTRACT
The introduction of MenAfriVac has significantly reduced group A meningococcal meningitis in the African meningitis belt, but epidemics caused by other groups such as C, W, Y and X (MenCWYX) remain a threat. To address this, a new multivalent meningococcal conjugate vaccine (MMCV) has been developed and pre-qualified by WHO. This study extends a previously established transmission dynamic model for MenA to include MenCWYX, enabling evaluation of the potential impact of MMCVs under various vaccination strategies. Using Burkina Faso as a case study, the model simulates mass campaigns targeting different age groups and routine vaccination through the Essential Programme on Immunization (EPI). The results indicate that campaigns targeting 1-29-year-olds are most effective in averting cases and delaying disease resurgence, while 1-19-year-old campaigns offer a resource-efficient alternative. Vaccine efficacy against carriage and the duration of protection significantly influence outcomes; greater efficacy (90% vs. 60%) and longer protection delay resurgence and reduce the number of cases. Routine- only vaccination demonstrates value in lower-risk settings, though it is less effective than combined strategies. Sensitivity analyses confirm the robustness of the ranking of strategies but highlight the importance of accurate estimates of vaccine efficacy and transmission parameters. The findings suggest that countries in the meningitis belt should integrate MMCVs into their immunisation programs, with high-risk countries prioritising catch-up campaigns for children and young adults. Despite data limitations and uncertainties, this model provides valuable insights for optimising vaccine rollout and highlights critical research needs, such as understanding vaccine effectiveness against carriage. These results support informed decision-making to sustain progress against meningitis and protect populations from future epidemics. MMCVs hold great promise in further reducing meningitis burden and approaching disease elimination in the region.
PMID:40880354 | DOI:10.1371/journal.pone.0330627
Veterinary Care Assistant [Temporary Cover]
Salary: £22,954 - £23,400 per annum (which is 94.79% of the full-time salary bracket of £24,215 - £24,685) + 15% Shift Allowance = c. £26,397 - £26,910 per annum.
This equates to an hourly rate of £12.70 to £12.94 plus a 15% shift allowance.
We have an exciting opportunity for someone to join us as a 24/7 Veterinary Care Assistant in our Small Animal Wing on a temporary basis. The referral hospital is a very fast-paced environment working with complex and seriously ill animals. The temporary role will begin as soon as possible from September 2025 and will continue until the November 2025.
The main objective of the role is to provide animal care to an excellent standard for the Queen's Veterinary School referral Hospital, to meet the needs of the service in the Small Animal Wing. The small animal wing consists of four dog, two cat, critical care, isolation wards housing an average of 15-25 patients during the overnight period and Theatre Suite.
You will play an important part in a primary care team working alongside nurses and other veterinary care assistants. Responsibilities will include cleaning and animal care duties in order to assist veterinary nurses in the inpatient area and in theatres.
In return, we offer an encouraging and nurturing environment and have a dedicated team of clinicians, nurses and veterinary care assistants who are committed to providing the best care for our patients.
Benefits
- Generous paid annual leave including bank holidays
- Defined benefit pension scheme
- Enhanced family friendly policies
- Access to a dedicated Personal and Professional Development team
- Wellness programme including Occupational Health team and Staff counselling
- Staff discount scheme including shopping vouchers
- Cycle to work scheme
- Travel to work loans
- Eye care voucher scheme
- Discounted gym membership
If you have any questions about this role please contact the Clinical HR Team by email: qvsh.hr@vet.cam.ac.uk
Further particulars for the role and information about the Department are available at: www.vet.cam.ac.uk
Click the 'Apply' button below to register an account with our recruitment system (if you have not already) and apply online.
Closing date: Sunday, 7th September 2025
Applications will be monitored regularly, and we may contact candidates prior to the closing date. Therefore, if you are interested, please submit your application as early as possible.
Please quote reference PP47031 on your application and in any correspondence about this vacancy.
The University actively supports equality, diversity and inclusion and encourages applications from all sections of society.
The University has a responsibility to ensure that all employees are eligible to live and work in the UK.
Cardiac lymphoma causing severe pulmonary stenosis in a cat
J Vet Cardiol. 2025 Jul 15;61:56-61. doi: 10.1016/j.jvc.2025.07.001. Online ahead of print.
ABSTRACT
A nine-year-old, domestic shorthair cat was referred for investigation of a suspected renal mass, polyuria, polydipsia, hyporexia and weight loss of one month's duration; no respiratory signs were reported. On presentation, the cat had marked respiratory effort. Thoracic auscultation revealed reduced heart and lung sounds bilaterally. Transthoracic echocardiography revealed a large pleural effusion and an extensive, homogenous mass within the right ventricular outflow tract, invading the pulmonary valve and causing severe infundibular and valvular pulmonary stenosis and right atrial and ventricular dilatation. The mass extended to, and infiltrated, the right ventricular free wall. Postmortem examination confirmed the presence of a mass associated with the pulmonary valve extending into the right ventricle and infiltrating the right ventricular and right atrial myocardium, resulting in severe infundibular and valvular pulmonary stenosis. Histopathology showed disseminated intermediate to large cell lymphoma affecting the kidney, myocardium, pulmonary valve, pancreas, diaphragm and adrenal glands.
PMID:40795482 | DOI:10.1016/j.jvc.2025.07.001
Multiscale Profiling of Nanoscale Metal-Organic Framework Biocompatibility and Immune Interactions
Adv Healthc Mater. 2025 Aug 7:e01809. doi: 10.1002/adhm.202501809. Online ahead of print.
ABSTRACT
The clinical translation of metal-organic frameworks (MOFs) - a promising class of porous materials for nanomedicine - is hindered by a poor understanding of their complex interactions with the immune system and in vivo immunotoxicity. To address this gap, a hierarchical "Safety-by-Design" pipeline is established and validated, integrating machine learning (ML) with ex vivo human blood studies and targeted in vivo models. This multi-stage workflow enables the systematic profiling of MOF immunotoxicity, de-risking their development. The power of this approach is demonstrated using four clinically relevant MOFs - NU-901, PCN-222, UiO-66, and ZIF-8 - revealing distinct, framework-dependent immune fingerprints. The initial in silico screening correctly flagged NU-901 and ZIF-8 as potential hazards to human health. These predictions are subsequently validated ex vivo, where NU-901 is confirmed to be selectively cytotoxic to CD14+ monocytes, and ZIF-8 is identified as a specific pro-inflammatory agent via IL-6 induction. In contrast, candidates predicted to be safe - UiO-66 and PCN-222 - demonstrated high biocompatibility ex vivo and advanced to in vivo studies, where they caused only minimal and transient immune activation. This study provides a validated, resource-efficient roadmap for preclinical immunotoxicity assessment, establishing a rational paradigm to accelerate the safe clinical translation of MOFs and other advanced nanomedicines.
PMID:40772350 | DOI:10.1002/adhm.202501809
Unwelcome neighbours: Tracking the transmission of Streptococcus equi in the United Kingdom horse population
Equine Vet J. 2025 Jul 20. doi: 10.1111/evj.14558. Online ahead of print.
ABSTRACT
BACKGROUND: Strangles (Streptococcus equi infection) remains endemic in the UK, with ~300 laboratory diagnoses annually. Sub-clinically infected long-term carriers are considered a key driver of endemicity. Analysing genomes of circulating strains could provide valuable transmission insights of this pathogen.
OBJECTIVES: To determine the population structure and diversity of UK S. equi isolates and to model transmission using epidemiological and whole genome sequencing data.
STUDY DESIGN: Retrospective cross-sectional epidemiological and genomic surveillance.
METHODS: A dated phylogenetic tree derived from 511 S. equi isolates collected from UK horses between 2015 and 2022 was reconstructed. Bayesian Analysis of Population Structure (BAPS) identified clusters of related genomes, while iGRAPH identified clusters of sequences appropriate for transmission analysis, performed using Transphylo.
RESULTS: BAPS identified nine groups, with 82% of strains clustering into two (McG-BAPS3, McG-BAPS5). A statistically significant association (p < 0.001) was found between the year of recovery and trends in the frequency of McG-BAPS groups, with McG-BAPS3 increasing and McG-BAPS5 decreasing in prevalence over the study period. Eight transmission clusters encompassing 64% of total sequences (n = 286/447) underwent analysis. Sixteen direct transmission pairs were identified; 10 were between horses from different UK regions. A transmission chain extending over a 6-month period was inferred from isolates from nine horses.
MAIN LIMITATIONS: Bacterial strains from sub-clinically infected carrier horses may be underrepresented due to data collection via positive laboratory diagnoses. Furthermore, a low sampling proportion relative to overall UK cases provided only a snapshot of broader, unsampled transmission events.
CONCLUSIONS: The rapid change in S. equi population structure indicates acutely infected/recently convalesced short-term carrier horses play a more influential role in transmission than long-term carriers. Our work provides novel insights to our understanding of S. equi transmission dynamics. Transmission of genetically related strains across diverse regions suggests a real-time sequence-based surveillance system could inform interventions to minimise transmission.
PMID:40684376 | DOI:10.1111/evj.14558
Global dissemination of npmA mediated pan-aminoglycoside resistance via a mobile genetic element in Gram-positive bacteria
Nat Commun. 2025 Jul 17;16(1):6360. doi: 10.1038/s41467-025-61152-y.
ABSTRACT
The npmA gene, encoding a 16S rRNA methyltransferase, confers resistance to all clinically available aminoglycosides, posing a significant threat to effective antibiotic therapy. We analyze 1,932,812 bacterial genomes to investigate the distribution and mobilization of npmA variants. npmA is not found in Gram-negative bacteria, where it was originally described, but is identified among Gram-positive bacteria, predominantly as the npmA2 variant in the globally distributed Clostridioides difficile ST11 lineage. We also detect npmA2 in two vancomycin-resistant Enterococcus faecium isolates from a Dutch hospital. Upon sequencing and phenotypic analysis, we determine that E. faecium isolates are pan-resistant to aminoglycosides. Genomic characterization links npmA2 to a composite transposon, Tn7734, which is integrated within a previously uncharacterized Integrative and Conjugative Element (ICE) Tn7740, present in both npmA2-carrying C. difficile and E. faecium clinical isolates. Tn7740-like, but not npmA2, appears across diverse taxa, including human microbiome members. Here, we show that Tn7740 likely facilitates cross-species npmA2 mobilization between these Gram-positive bacteria and emphasize the risk of mobile genetic elements transferring pan-aminoglycoside resistance between clinically important bacterial pathogens.
PMID:40675954 | DOI:10.1038/s41467-025-61152-y
Global diversity of soil-transmitted helminths reveals population-biased genetic variation that impacts diagnostic targets
Nat Commun. 2025 Jul 10;16(1):6374. doi: 10.1038/s41467-025-61687-0.
ABSTRACT
Soil-transmitted helminths (STHs) are intestinal parasites that affect over a billion people worldwide. STH control relies on microscopy-based diagnostics to monitor parasite prevalence and enable post-treatment surveillance; however, molecular diagnostics are rapidly being developed due to increased sensitivity, particularly in low-STH-prevalence settings. The genetic diversity of helminths and its potential impact on molecular diagnostics remain unclear. Using low-coverage genome sequencing, we assess the genetics of STHs within worm, faecal, and purified egg samples from 27 countries, identifying differences in the genetic connectivity and diversity of STH-positive samples across regions and cryptic diversity between closely related human- and pig-infective species. We define substantial copy number and sequence variants in current diagnostic target regions and validate the impact of genetic variation on qPCR diagnostics using in vitro assays. Our study provides insights into the diversity and genomic epidemiology of STHs, highlighting both the challenges and opportunities for developing molecular diagnostics needed to support STH control efforts.
PMID:40640199 | DOI:10.1038/s41467-025-61687-0
Large-scale DNA study maps 37,000 years of human disease history
A new study suggests that our ancestors’ close cohabitation with domesticated animals and large-scale migrations played a key role in the spread of infectious diseases.
The team, led by Professor Eske Willerslev at the Universities of Cambridge and Copenhagen, recovered ancient DNA from 214 known human pathogens in prehistoric humans from Eurasia.
They found that the earliest evidence of zoonotic diseases – illnesses transmitted from animals to humans, like COVID in recent times – dates back to around 6,500 years ago, with these diseases becoming more widespread approximately 5,000 years ago.
The study detected the world’s oldest genetic trace of the plague bacterium, Yersinia pestis, in a 5,500-year-old sample. The plague is estimated to have killed between one-quarter and one-half of Europe’s population during the Middle Ages.
In addition, the researchers found traces of many other diseases including:
Malaria (Plasmodium vivax) – 4,200 years ago
Leprosy (Mycobacterium leprae) – 1,400 years ago
Hepatitis B virus – 9,800 years ago
Diphtheria (Corynebacterium diphtheriae) – 11,100 years ago
This is the largest study to date on the history of infectious diseases and is published today in the journal Nature.
The researchers analysed DNA from over 1,300 prehistoric humans, some up to 37,000 years old. The ancient bones and teeth have provided a unique insight into the development of diseases caused by bacteria, viruses, and parasites.
“We’ve long suspected that the transition to farming and animal husbandry opened the door to a new era of disease – now DNA shows us that it happened at least 6,500 years ago,” said Willerslev.
He added: “These infections didn’t just cause illness – they may have contributed to population collapse, migration, and genetic adaptation.”
The significant increase in the incidence of zoonoses around 5,000 years ago coincides with a migration to north-western Europe from the Pontic Steppe – that is from parts of present-day Ukraine, south-western Russia and western Kazakhstan. The people embarking on this migration – and who to a large extent passed on the genetic profile found among people in north-western Europe today – belonged to the Yamnaya herders.
The findings could be significant for the development of vaccines and for understanding how diseases arise and mutate over time.
“If we understand what happened in the past, it can help us prepare for the future. Many of the newly emerging infectious diseases are predicted to originate from animals,” said Associate Professor Martin Sikora at the University of Copenhagen, and first author of the report.
Willerslev added: “Mutations that were successful in the past are likely to reappear. This knowledge is important for future vaccines, as it allows us to test whether current vaccines provide sufficient coverage or whether new ones need to be developed due to mutations.”
The sample material was primarily provided by museums in Europe and Asia. The samples were partly extracted from teeth, where the enamel acts as a lid that can protect the DNA against degradation as a result of the ravages of time. The rest of the DNA was primarily extracted from petrosa bones - the hardest bone in humans - located on the inside of the skull.
The research was funded by the Lundbeck Foundation.
ReferenceSikora, M. et al: ‘The spatiotemporal distribution of human pathogens in ancient Eurasia.’ Nature, July 2025. DOI: 10.1038/s41586-025-09192-8
Adapted from a press release by the University of Copenhagen.
Researchers have mapped the spread of infectious diseases in humans across millennia, to reveal how human-animal interactions permanently transformed our health today.
We’ve long suspected that the transition to farming and animal husbandry opened the door to a new era of disease – now DNA shows us that it happened at least 6,500 years agoEske WillerslevMarie Louise JørkovLate Neolithic skull from Madesø
The text in this work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. Images, including our videos, are Copyright ©University of Cambridge and licensors/contributors as identified. All rights reserved. We make our image and video content available in a number of ways – on our main website under its Terms and conditions, and on a range of channels including social media that permit your use and sharing of our content under their respective Terms.
The spatiotemporal distribution of human pathogens in ancient Eurasia
Nature. 2025 Jul 9. doi: 10.1038/s41586-025-09192-8. Online ahead of print.
ABSTRACT
Infectious diseases have had devastating effects on human populations throughout history, but important questions about their origins and past dynamics remain1. To create an archaeogenetic-based spatiotemporal map of human pathogens, we screened shotgun-sequencing data from 1,313 ancient humans covering 37,000 years of Eurasian history. We demonstrate the widespread presence of ancient bacterial, viral and parasite DNA, identifying 5,486 individual hits against 492 species from 136 genera. Among those hits, 3,384 involve known human pathogens2, many of which had not previously been identified in ancient human remains. Grouping the ancient microbial species according to their likely reservoir and type of transmission, we find that most groups are identified throughout the entire sampling period. Zoonotic pathogens are only detected from around 6,500 years ago, peaking roughly 5,000 years ago, coinciding with the widespread domestication of livestock3. Our findings provide direct evidence that this lifestyle change resulted in an increased infectious disease burden. They also indicate that the spread of these pathogens increased substantially during subsequent millennia, coinciding with the pastoralist migrations from the Eurasian Steppe4,5.
PMID:40634616 | DOI:10.1038/s41586-025-09192-8
Enrichment of Helminth Mitochondrial Genomes From Faecal Samples Using Hybridisation Capture
Mol Ecol Resour. 2025 Jul 9:e70005. doi: 10.1111/1755-0998.70005. Online ahead of print.
ABSTRACT
New approaches are urgently needed to enrich rare or low-abundant DNA in complex samples. Soil-transmitted helminths (STHs) inhabit heterogeneous environments, including the gastrointestinal tract of their host as adults and are excreted as eggs and larvae in faeces, complicating our understanding of their biology and the use of genetic tools for species monitoring and population tracking. We have developed a hybridisation capture approach to enrich mitochondrial genome sequences of two STH species, the roundworm Ascaris lumbricoides and whipworm Trichuris trichiura, from extracted DNA from faecal material and worm specimens. Employing ~1000 targeted probes, we achieved > 6000 and > 12,000 fold enrichment for A. lumbricoides and T. trichiura, respectively, relative to direct whole genome shotgun (WGS) sequencing. Sequencing coverage was highly concordant with probe targets and correlated with the number of eggs per gram (EPG) of parasites present, from which DNA from as few as 336 EPG for Ascaris and 48 EPG for Trichuris were efficiently captured and sufficient to provide effective mitochondrial genome data. Finally, allele frequencies were highly concordant between WGS and hybridisation capture, suggesting little genetic information is lost with additional sample processing required for enrichment. Our hybridisation capture design and approach enable sensitive and flexible STH mitochondrial genome sampling from faecal DNA extracts and pave the way for broader hybridisation capture-based genome-wide applications and molecular epidemiology studies of STHs.
PMID:40631469 | DOI:10.1111/1755-0998.70005
- 1
- 2