{kind=link}
Immunogenicity of a trivalent haemorrhagic fever vaccine candidate against Sudan virus, Marburg virus and Lassa virus in an mpox vaccine
J Gen Virol. 2025 Oct;106(10). doi: 10.1099/jgv.0.002157.
ABSTRACT
A multivalent vaccine targeting high-consequence infectious diseases in Sub-Saharan Africa (SSA), which are linked to high mortality, morbidity and overlapping clinical manifestations, would significantly improve health security and economic stability in this region. Trivalent vector vaccines were devised to deliver digitally optimized versions of Orthoebolavirus, Orthomarburgvirus glycoproteins (GPs) and a Lassa mammarenavirus (LASV) nucleoprotein (NP) by a single Modified Vaccinia Ankara (MVA) known to protect against mpox virus (MPXV) along with a matched DNA vaccine. Three immunizations in mice and Hartley guinea pigs with MVA only or a DNA prime followed by two MVA administrations induced comparable levels of binding antibodies and LASV-specific T-cells, respectively. While DNA priming mitigated MVA-specific antibody responses, GP- and NP-specific antibodies developed already after a single MVA vaccination. Although a post-outbreak Ebola virus vaccine is available, outbreaks by other filoviruses, annual LASV epidemics and increased incidence of MPXV infections support the rationale for an MVA-based trivalent haemorrhagic fever vaccine for endemic and high-risk human populations in SSA.
PMID:41051941 | DOI:10.1099/jgv.0.002157
A Single Institution Retrospective Comparison of Two Radiotherapy Protocols for the Palliative Treatment of Canine Nasal Carcinoma
Vet Radiol Ultrasound. 2025 Nov;66(6):e70097. doi: 10.1111/vru.70097.
ABSTRACT
Optimal radiation protocols for canine nasal carcinoma are not established. Co-morbidities, access, and owner compliance can influence scheduling. Between 2015 and 2022, two radiotherapy protocols were used in the palliative treatment of canine nasal carcinoma at a single institution. Group A comprised 17 cases receiving 40 Gy in ten 4 Gy fractions delivered Monday, Wednesday, and Friday. Epistaxis was present in 11/17 (65%) cases. Median survival time (MST) was 298 days (95% CI: 163.54-432.45); progression-free survival was 173 days (95% CI: 117.87-228.12). Group B comprised 24 cases receiving 36 Gy in six 6 Gy fractions delivered Monday and Friday. Epistaxis was present in 20/24 (83%) cases. MST was 375 days (95% CI: 240.73-509.27); progression-free survival was 243 days (95% CI: 138.42-347.58). Dogs with Adams Stage 1 disease had the longest median overall (593 days) and progression-free survival (609 days). Four cases each received additional radiation treatment and/or toceranib at relapse. Palliative radiation therapy achieved control of clinical signs in the majority of cases, with an overall response rate of 100% (Group A) and 96% (Group B). In a multivariate Cox regression model with backwards elimination, when cases were stratified for tumor stage, neither the presence of epistaxis nor treatment (6 vs. 10 fractions) was independently associated with significant improvements in survival. Epistaxis at presentation did not appear to influence survival. These results indicate that palliative radiation therapy is highly effective in controlling clinical signs associated with nasal carcinoma. Increasing fractionation may have a limited effect on survival outcome or toxicity in the palliative setting.
PMID:41047698 | DOI:10.1111/vru.70097
Editorial: Strategies for mitigating zoonotic influenza outbreaks: a comprehensive preparedness approach
Front Public Health. 2025 Sep 3;13:1685224. doi: 10.3389/fpubh.2025.1685224. eCollection 2025.
NO ABSTRACT
PMID:40969630 | PMC:PMC12441160 | DOI:10.3389/fpubh.2025.1685224
Antimicrobial use differs between general practice and referral settings in United Kingdom companion animals: a 12-month prospective study
Am J Vet Res. 2025 Sep 12:1-10. doi: 10.2460/ajvr.25.06.0229. Online ahead of print.
ABSTRACT
OBJECTIVE: To characterize the use of antimicrobials and frequency of microbial identification testing in cases seen by companion animal general practitioners and a single United Kingdom (UK) internal medicine referral service.
METHODS: This was a prospective, observational study of animals referred to a single UK-based internal medicine referral service over a 12-month period. Information recorded at the time of presentation included presenting complaint, current medications, and whether a microbial identification test had been performed. At discharge, the same information was recorded again alongside the final diagnosis.
RESULTS: 516 dogs and cats were enrolled, and 22.9% of cases were receiving antimicrobials at the time of presentation compared to 22.0% of cases at the time of discharge. Cases receiving antimicrobials at admittance were 2.7 (95% CI, 1.7 to 4.3) times more likely to be discharged on antimicrobials. Postreferral respiratory and urinary cases were 7.3 times more likely (OR, 7.3; 95% CI, 3.6 to 15.0) and 4.2 times more likely (OR, 4.2; 95% CI, 1.9 to 9.4), respectively, to be discharged on antimicrobials. Bacterial culture was more commonly performed within the referral center (72.1%) than prior to referral (16.9%).
CONCLUSIONS: Bacterial culture is performed more frequently by referral internal medicine clinicians than UK general practitioners. There are differences in the distribution of cases prescribed antimicrobials between these 2 groups of veterinarians.
CLINICAL RELEVANCE: This study provides information on antimicrobial prescribing patterns in companion animals across both general practice and referral settings in the UK. It highlights areas for antimicrobial stewardship improvement as well as identifies factors associated with antimicrobial prescription.
PMID:40939627 | DOI:10.2460/ajvr.25.06.0229
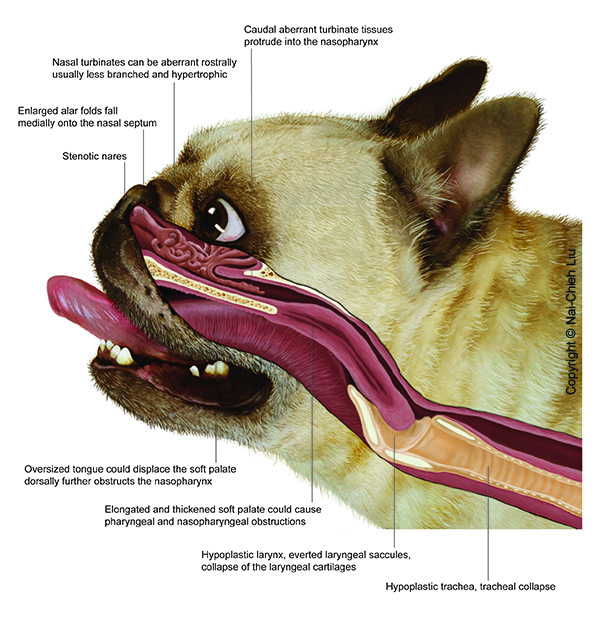
Pterygoid bone malformation and its limitations on the effectiveness of brachycephalic airway corrective surgery in brachycephalic dogs
J Small Anim Pract. 2025 Sep 11. doi: 10.1111/jsap.70028. Online ahead of print.
ABSTRACT
OBJECTIVES: This study aimed to examine the association between pterygoid bone medialisation and treatment outcomes after upper airway surgery in three brachycephalic breeds.
MATERIALS AND METHODS: Dogs that underwent CT of the head followed by routine surgery for brachycephalic obstructive airway syndrome were recruited in this study. Measurements obtained from the CT images included the width of the basisphenoid bone, interpterygoid distance and cross-sectional area of the nasopharynx. A ratio of width of the basisphenoid bone to interpterygoid distance allowed quantification of pterygoid bone medialisation. Pearson's correlations were calculated to assess the relationship between width of the basisphenoid bone: interpterygoid distance and cross-sectional area of the nasopharynx. Additionally, width of the basisphenoid bone: interpterygoid distance was compared across breeds, as well as between dogs with good and poor surgical outcomes (post-operative brachycephalic obstructive airway syndrome index ≥50%).
RESULTS: One hundred and forty-four brachycephalic dogs (47 Pugs, 64 French Bulldogs and 33 Bulldogs) and 30 non-brachycephalic controls were included in the analysis. The width of the basisphenoid bone: interpterygoid distance ratio in brachycephalic dogs was significantly higher (1.982 ± 0.379) than that of controls (1.646 ± 0.239, P < 0.001). A negative correlation was observed between width of the basisphenoid bone: interpterygoid distance and cross-sectional area of the nasopharynx in Pugs (ρ = -0.29, P = 0.048), French Bulldogs (ρ = -0.47, P < 0.001), Bulldogs (ρ = -0.71, P < 0.001) and controls (ρ = -0.55, P = 0.002). French Bulldogs with poor surgical outcomes exhibited a significantly higher width of the basisphenoid bone: interpterygoid distance (2.366 ± 0.327) than those with good surgical outcomes (1.813 ± 0.271, P < 0.0001).
CLINICAL SIGNIFICANCE: Pterygoid bone medialisation is associated with nasopharyngeal narrowing, which limits the effectiveness of surgical interventions in brachycephalic obstructive airway syndrome in affected French Bulldogs. As there are no surgical options currently reported to address this condition, these findings are important in guiding clinicians in providing prognostic information to owners during elective brachycephalic obstructive airway syndrome surgery.
PMID:40935636 | DOI:10.1111/jsap.70028
Use of image-guided robotic-assisted drilling for transcondylar screw placement in the canine humerus
Vet Surg. 2025 Sep 5. doi: 10.1111/vsu.70010. Online ahead of print.
ABSTRACT
OBJECTIVE: To determine if a novel robotic system has comparable positional and angular accuracy to that achievable with patient-specific guides (PSG) when used for transcondylar screw (TCS) placement in the canine humerus.
STUDY DESIGN: Experimental laboratory study.
SAMPLE POPULATION: A total of 32 synthetic humeral models (16 per group).
METHODS: Bone models were three-dimensional (3D)-printed and drilled with the aid of a custom PSG or with the assistance of an image-guided surgical robot. A 2.5-mm hole was drilled medial to lateral and the entry point, exit point and angular trajectory of the drill hole were measured on postoperative computed tomography (CT) scans. Absolute differences between planned and actual positions and trajectories were compared between PSG and Robot groups.
RESULTS: None of the drill holes in this study violated the articular surface of the humerus. Entry point positioning was significantly more accurate in the PSG group, but drill hole trajectories (angulation) were more accurate in the Robot group. Exit point positioning was similar in the two groups.
CONCLUSION: Robotic assistance enables safe placement of drill holes for TCS. PSG enable more accurate drill entry, but robotic assistance allows for more accurate overall drill hole trajectory.
CLINICAL SIGNIFICANCE: Robotic assistance allows for accurate and safe drilling of screw holes for TCS placement in the humerus. The robotic procedure allows for a more limited surgical exposure, but the technical feasibility and outcomes associated with this approach should now be evaluated in cadavers before moving to clinical evaluation in live patients.
PMID:40913298 | DOI:10.1111/vsu.70010
Use of ultrasound to estimate tracheal diameter in unclipped brachycephalic dogs: A pilot study
Vet Rec Open. 2025 Sep 2;12(2):e70018. doi: 10.1002/vro2.70018. eCollection 2025 Dec.
ABSTRACT
BACKGROUND: A significant contributory factor to the brachycephalic obstructive airway syndrome index of bulldogs is the diameter of their tracheas. Bulldogs are predisposed to tracheal hypoplasia. A non-invasive, financially reasonable and widely accessible screening test for tracheal diameter would be useful for assessing the most appropriate dogs to breed from within bulldog populations and may help in predicting results following upper airway surgery.
METHOD: A prospective method comparison study involving 10 client-owned brachycephalic dogs was conducted. Each patient underwent tracheal ultrasonography using a handheld ultrasound device (Butterfly IQ+) followed by extubated computed tomography (CT) scanning of the head and neck. Ultrasonographic tracheal measurements were compared with CT tracheal measurements and analysed for agreement, accuracy, and inter-observer and intra-observer repeatability.
RESULTS: Bland‒Altman analysis showed there was good agreement between the ultrasound and CT tracheal measurements; however, the 95% limits of agreement were wide (+0.43 and ‒0.29 cm), indicating that the ultrasound measurements lacked a high level of accuracy. Ultrasound in general overestimated the tracheal diameter by 0.07 cm (p < 0.05). Intra-observer repeatability (mean range: 0.12 cm, average coefficient of variation [COV]: observer one;7.36%, observer two;5.53%, observer three;6.10%) was more consistent than inter-observer repeatability (mean range: 0.26 cm, average COV: 8.47%).
CONCLUSION: The accuracy of tracheal diameter measurements using an affordable handheld ultrasound device in unclipped brachycephalic patients was relatively low. However, technique modifications may significantly improve results, and further investigation is warranted to explore the utility of this method as a screening tool for tracheal hypoplasia in bulldog populations.
PMID:40904891 | PMC:PMC12403226 | DOI:10.1002/vro2.70018
Modelling of strategies for the introduction and routine use of multivalent meningococcal conjugate vaccines (MMCVs) in the African meningitis belt
PLoS One. 2025 Aug 29;20(8):e0330627. doi: 10.1371/journal.pone.0330627. eCollection 2025.
ABSTRACT
The introduction of MenAfriVac has significantly reduced group A meningococcal meningitis in the African meningitis belt, but epidemics caused by other groups such as C, W, Y and X (MenCWYX) remain a threat. To address this, a new multivalent meningococcal conjugate vaccine (MMCV) has been developed and pre-qualified by WHO. This study extends a previously established transmission dynamic model for MenA to include MenCWYX, enabling evaluation of the potential impact of MMCVs under various vaccination strategies. Using Burkina Faso as a case study, the model simulates mass campaigns targeting different age groups and routine vaccination through the Essential Programme on Immunization (EPI). The results indicate that campaigns targeting 1-29-year-olds are most effective in averting cases and delaying disease resurgence, while 1-19-year-old campaigns offer a resource-efficient alternative. Vaccine efficacy against carriage and the duration of protection significantly influence outcomes; greater efficacy (90% vs. 60%) and longer protection delay resurgence and reduce the number of cases. Routine- only vaccination demonstrates value in lower-risk settings, though it is less effective than combined strategies. Sensitivity analyses confirm the robustness of the ranking of strategies but highlight the importance of accurate estimates of vaccine efficacy and transmission parameters. The findings suggest that countries in the meningitis belt should integrate MMCVs into their immunisation programs, with high-risk countries prioritising catch-up campaigns for children and young adults. Despite data limitations and uncertainties, this model provides valuable insights for optimising vaccine rollout and highlights critical research needs, such as understanding vaccine effectiveness against carriage. These results support informed decision-making to sustain progress against meningitis and protect populations from future epidemics. MMCVs hold great promise in further reducing meningitis burden and approaching disease elimination in the region.
PMID:40880354 | DOI:10.1371/journal.pone.0330627
Cardiac lymphoma causing severe pulmonary stenosis in a cat
J Vet Cardiol. 2025 Jul 15;61:56-61. doi: 10.1016/j.jvc.2025.07.001. Online ahead of print.
ABSTRACT
A nine-year-old, domestic shorthair cat was referred for investigation of a suspected renal mass, polyuria, polydipsia, hyporexia and weight loss of one month's duration; no respiratory signs were reported. On presentation, the cat had marked respiratory effort. Thoracic auscultation revealed reduced heart and lung sounds bilaterally. Transthoracic echocardiography revealed a large pleural effusion and an extensive, homogenous mass within the right ventricular outflow tract, invading the pulmonary valve and causing severe infundibular and valvular pulmonary stenosis and right atrial and ventricular dilatation. The mass extended to, and infiltrated, the right ventricular free wall. Postmortem examination confirmed the presence of a mass associated with the pulmonary valve extending into the right ventricle and infiltrating the right ventricular and right atrial myocardium, resulting in severe infundibular and valvular pulmonary stenosis. Histopathology showed disseminated intermediate to large cell lymphoma affecting the kidney, myocardium, pulmonary valve, pancreas, diaphragm and adrenal glands.
PMID:40795482 | DOI:10.1016/j.jvc.2025.07.001
Multiscale Profiling of Nanoscale Metal-Organic Framework Biocompatibility and Immune Interactions
Adv Healthc Mater. 2025 Aug 7:e01809. doi: 10.1002/adhm.202501809. Online ahead of print.
ABSTRACT
The clinical translation of metal-organic frameworks (MOFs) - a promising class of porous materials for nanomedicine - is hindered by a poor understanding of their complex interactions with the immune system and in vivo immunotoxicity. To address this gap, a hierarchical "Safety-by-Design" pipeline is established and validated, integrating machine learning (ML) with ex vivo human blood studies and targeted in vivo models. This multi-stage workflow enables the systematic profiling of MOF immunotoxicity, de-risking their development. The power of this approach is demonstrated using four clinically relevant MOFs - NU-901, PCN-222, UiO-66, and ZIF-8 - revealing distinct, framework-dependent immune fingerprints. The initial in silico screening correctly flagged NU-901 and ZIF-8 as potential hazards to human health. These predictions are subsequently validated ex vivo, where NU-901 is confirmed to be selectively cytotoxic to CD14+ monocytes, and ZIF-8 is identified as a specific pro-inflammatory agent via IL-6 induction. In contrast, candidates predicted to be safe - UiO-66 and PCN-222 - demonstrated high biocompatibility ex vivo and advanced to in vivo studies, where they caused only minimal and transient immune activation. This study provides a validated, resource-efficient roadmap for preclinical immunotoxicity assessment, establishing a rational paradigm to accelerate the safe clinical translation of MOFs and other advanced nanomedicines.
PMID:40772350 | DOI:10.1002/adhm.202501809
Unwelcome neighbours: Tracking the transmission of Streptococcus equi in the United Kingdom horse population
Equine Vet J. 2025 Jul 20. doi: 10.1111/evj.14558. Online ahead of print.
ABSTRACT
BACKGROUND: Strangles (Streptococcus equi infection) remains endemic in the UK, with ~300 laboratory diagnoses annually. Sub-clinically infected long-term carriers are considered a key driver of endemicity. Analysing genomes of circulating strains could provide valuable transmission insights of this pathogen.
OBJECTIVES: To determine the population structure and diversity of UK S. equi isolates and to model transmission using epidemiological and whole genome sequencing data.
STUDY DESIGN: Retrospective cross-sectional epidemiological and genomic surveillance.
METHODS: A dated phylogenetic tree derived from 511 S. equi isolates collected from UK horses between 2015 and 2022 was reconstructed. Bayesian Analysis of Population Structure (BAPS) identified clusters of related genomes, while iGRAPH identified clusters of sequences appropriate for transmission analysis, performed using Transphylo.
RESULTS: BAPS identified nine groups, with 82% of strains clustering into two (McG-BAPS3, McG-BAPS5). A statistically significant association (p < 0.001) was found between the year of recovery and trends in the frequency of McG-BAPS groups, with McG-BAPS3 increasing and McG-BAPS5 decreasing in prevalence over the study period. Eight transmission clusters encompassing 64% of total sequences (n = 286/447) underwent analysis. Sixteen direct transmission pairs were identified; 10 were between horses from different UK regions. A transmission chain extending over a 6-month period was inferred from isolates from nine horses.
MAIN LIMITATIONS: Bacterial strains from sub-clinically infected carrier horses may be underrepresented due to data collection via positive laboratory diagnoses. Furthermore, a low sampling proportion relative to overall UK cases provided only a snapshot of broader, unsampled transmission events.
CONCLUSIONS: The rapid change in S. equi population structure indicates acutely infected/recently convalesced short-term carrier horses play a more influential role in transmission than long-term carriers. Our work provides novel insights to our understanding of S. equi transmission dynamics. Transmission of genetically related strains across diverse regions suggests a real-time sequence-based surveillance system could inform interventions to minimise transmission.
PMID:40684376 | DOI:10.1111/evj.14558
Global dissemination of npmA mediated pan-aminoglycoside resistance via a mobile genetic element in Gram-positive bacteria
Nat Commun. 2025 Jul 17;16(1):6360. doi: 10.1038/s41467-025-61152-y.
ABSTRACT
The npmA gene, encoding a 16S rRNA methyltransferase, confers resistance to all clinically available aminoglycosides, posing a significant threat to effective antibiotic therapy. We analyze 1,932,812 bacterial genomes to investigate the distribution and mobilization of npmA variants. npmA is not found in Gram-negative bacteria, where it was originally described, but is identified among Gram-positive bacteria, predominantly as the npmA2 variant in the globally distributed Clostridioides difficile ST11 lineage. We also detect npmA2 in two vancomycin-resistant Enterococcus faecium isolates from a Dutch hospital. Upon sequencing and phenotypic analysis, we determine that E. faecium isolates are pan-resistant to aminoglycosides. Genomic characterization links npmA2 to a composite transposon, Tn7734, which is integrated within a previously uncharacterized Integrative and Conjugative Element (ICE) Tn7740, present in both npmA2-carrying C. difficile and E. faecium clinical isolates. Tn7740-like, but not npmA2, appears across diverse taxa, including human microbiome members. Here, we show that Tn7740 likely facilitates cross-species npmA2 mobilization between these Gram-positive bacteria and emphasize the risk of mobile genetic elements transferring pan-aminoglycoside resistance between clinically important bacterial pathogens.
PMID:40675954 | DOI:10.1038/s41467-025-61152-y
Global diversity of soil-transmitted helminths reveals population-biased genetic variation that impacts diagnostic targets
Nat Commun. 2025 Jul 10;16(1):6374. doi: 10.1038/s41467-025-61687-0.
ABSTRACT
Soil-transmitted helminths (STHs) are intestinal parasites that affect over a billion people worldwide. STH control relies on microscopy-based diagnostics to monitor parasite prevalence and enable post-treatment surveillance; however, molecular diagnostics are rapidly being developed due to increased sensitivity, particularly in low-STH-prevalence settings. The genetic diversity of helminths and its potential impact on molecular diagnostics remain unclear. Using low-coverage genome sequencing, we assess the genetics of STHs within worm, faecal, and purified egg samples from 27 countries, identifying differences in the genetic connectivity and diversity of STH-positive samples across regions and cryptic diversity between closely related human- and pig-infective species. We define substantial copy number and sequence variants in current diagnostic target regions and validate the impact of genetic variation on qPCR diagnostics using in vitro assays. Our study provides insights into the diversity and genomic epidemiology of STHs, highlighting both the challenges and opportunities for developing molecular diagnostics needed to support STH control efforts.
PMID:40640199 | DOI:10.1038/s41467-025-61687-0
The spatiotemporal distribution of human pathogens in ancient Eurasia
Nature. 2025 Jul 9. doi: 10.1038/s41586-025-09192-8. Online ahead of print.
ABSTRACT
Infectious diseases have had devastating effects on human populations throughout history, but important questions about their origins and past dynamics remain1. To create an archaeogenetic-based spatiotemporal map of human pathogens, we screened shotgun-sequencing data from 1,313 ancient humans covering 37,000 years of Eurasian history. We demonstrate the widespread presence of ancient bacterial, viral and parasite DNA, identifying 5,486 individual hits against 492 species from 136 genera. Among those hits, 3,384 involve known human pathogens2, many of which had not previously been identified in ancient human remains. Grouping the ancient microbial species according to their likely reservoir and type of transmission, we find that most groups are identified throughout the entire sampling period. Zoonotic pathogens are only detected from around 6,500 years ago, peaking roughly 5,000 years ago, coinciding with the widespread domestication of livestock3. Our findings provide direct evidence that this lifestyle change resulted in an increased infectious disease burden. They also indicate that the spread of these pathogens increased substantially during subsequent millennia, coinciding with the pastoralist migrations from the Eurasian Steppe4,5.
PMID:40634616 | DOI:10.1038/s41586-025-09192-8
Enrichment of Helminth Mitochondrial Genomes From Faecal Samples Using Hybridisation Capture
Mol Ecol Resour. 2025 Jul 9:e70005. doi: 10.1111/1755-0998.70005. Online ahead of print.
ABSTRACT
New approaches are urgently needed to enrich rare or low-abundant DNA in complex samples. Soil-transmitted helminths (STHs) inhabit heterogeneous environments, including the gastrointestinal tract of their host as adults and are excreted as eggs and larvae in faeces, complicating our understanding of their biology and the use of genetic tools for species monitoring and population tracking. We have developed a hybridisation capture approach to enrich mitochondrial genome sequences of two STH species, the roundworm Ascaris lumbricoides and whipworm Trichuris trichiura, from extracted DNA from faecal material and worm specimens. Employing ~1000 targeted probes, we achieved > 6000 and > 12,000 fold enrichment for A. lumbricoides and T. trichiura, respectively, relative to direct whole genome shotgun (WGS) sequencing. Sequencing coverage was highly concordant with probe targets and correlated with the number of eggs per gram (EPG) of parasites present, from which DNA from as few as 336 EPG for Ascaris and 48 EPG for Trichuris were efficiently captured and sufficient to provide effective mitochondrial genome data. Finally, allele frequencies were highly concordant between WGS and hybridisation capture, suggesting little genetic information is lost with additional sample processing required for enrichment. Our hybridisation capture design and approach enable sensitive and flexible STH mitochondrial genome sampling from faecal DNA extracts and pave the way for broader hybridisation capture-based genome-wide applications and molecular epidemiology studies of STHs.
PMID:40631469 | DOI:10.1111/1755-0998.70005
Discovery of glycerol phosphate and an immunogenic glycan motif in rhamnose-rich polysaccharides of Streptococcus uberis
Vet Res. 2025 Jul 7;56(1):139. doi: 10.1186/s13567-025-01574-0.
ABSTRACT
Streptococcus uberis is a causative pathogen of bovine mastitis with high genetic diversity. Rhamnose-rich polysaccharides (RPS) are abundant surface structures covalently anchored to peptidoglycan and represent promising vaccine candidates for several streptococcal pathogens. It was previously reported that the RPS of S. uberis strain 233 is composed of a repeating → 2)-α-L-Rhap-(1 → 3)-α-L-Rhap-(1 → disaccharide backbone decorated with α-D-Glcp side-chains. In this study, we identified a hitherto unknown glycerol phosphate (GroP) modification at the 6-OH of the Glc residue in S. uberis 233 RPS using nuclear magnetic resonance analysis. Comparative genomic analysis of 592 S. uberis genomes revealed significant diversity in the RPS biosynthesis gene cluster with six major RPS genotypes. RPS genotypes 1-4, representing 97.5% of the analyzed strains, all contained the rhamnan backbone biosynthesis genes shared between several streptococcal species, as well as a putative GroP transferase gene. Using rhamnan-reactive immune serum, we further demonstrated that rhamnan is a conserved and accessible glycan motif in S. uberis RPS genotype 1 and 2 strains, but this motif is inferred to be shielded by side-chains in genotype 4 strains. Importantly, experiments with sera from cattle, challenged intramammarily with S. uberis, revealed that the rhamnan backbone of S. uberis RPS is an immunogenic glycan motif and remained accessible to bovine IgG antibodies in the presence of single residue RPS side-chains. Overall, this study suggests that S. uberis RPS are modified with GroP and reports that RPS in most strains contain a conserved, immunogenic and antibody accessible rhamnan glycan motif.
PMID:40624561 | DOI:10.1186/s13567-025-01574-0
Usefulness of cerebrospinal fluid analysis in dogs and cats with suspected intracranial disease and normal magnetic resonance imaging
Front Vet Sci. 2025 Jun 20;12:1583988. doi: 10.3389/fvets.2025.1583988. eCollection 2025.
ABSTRACT
Cerebrospinal fluid (CSF) analysis is a common diagnostic tool in the investigation of neurological presentations. Whether its routine use after every brain magnetic resonance imaging (MRI) is warranted is debated amongst clinicians, and its usefulness after a normal MRI has not yet been examined. To investigate whether CSF analysis affected the final diagnosis in dogs and cats with suspected intracranial disease in the presence of unremarkable magnetic resonance imaging (MRI), clinical, imaging and laboratory records of dogs and cats with suspected intracranial disease, unremarkable MRI and CSF analysis were reviewed in this multi-center retrospective study. Of 593 animals, (533 dogs and 60 cats), 17 dogs (3%) had abnormal CSF, nine of these demonstrating pleocytosis (with or without elevated microprotein) and eight showing hyperproteinorrachia alone. In only five of these dogs (0.8% of the total cohort) was the final diagnosis and/or treatment meaningfully affected by CSF findings: three diagnosed with inflammatory brain conditions and two had undetermined diagnoses, with corticosteroids initiated following abnormal CSF results. No cats in this population had an abnormal CSF. All dogs with a diagnosis based on abnormal CSF results had an abnormal neurological examination. In this population, CSF analysis was unlikely to reveal an undiagnosed intracranial condition following an unremarkable brain MRI, particularly in dogs presenting with a normal neurological examination. In dogs presenting with an abnormal neurological examination or a high suspicion of inflammatory disease, CSF evaluation following normal MRI is more likely to be diagnostically valuable.
PMID:40621502 | PMC:PMC12226868 | DOI:10.3389/fvets.2025.1583988
Immunocompetent cell targeting by food-additive titanium dioxide
Nat Commun. 2025 Jul 4;16(1):6067. doi: 10.1038/s41467-025-60248-9.
ABSTRACT
Food-grade titanium dioxide (fgTiO2) is a bio-persistent particle under intense regulatory scrutiny. Yet paradoxically, the only known cell reservoirs for fgTiO2 are graveyard intestinal pigment cells which are metabolically and immunologically quiescent. Here we identify immunocompetent cell targets of fgTiO2 in humans, most notably in the subepithelial dome region of intestinal Peyer's patches. Using multimodal microscopies with single-particle detection and per-cell / vesicle image analysis we achieve correlative dosimetry, quantitatively recapitulating human cellular exposures in the ileum of mice fed a fgTiO2-containing diet. Epithelial microfold cells selectively funnel fgTiO2 into LysoMac and LysoDC cells with ensuing accumulation. Notwithstanding, proximity extension analyses for 92 protein targets reveal no measureable perturbation of cell signalling pathways. When chased with oral ΔaroA-Salmonella, pro-inflammatory signalling is confirmed, but no augmentation by fgTiO2 is revealed despite marked same-cell loading. Interestingly, Salmonella causes the fgTiO2-recipient cells to migrate within the patch and, sporadically, to be identified in the lamina propria, thereby fully recreating the intestinal tissue distribution of fgTiO2 in humans. Immunocompetent cells that accumulate fgTiO2 in vivo are now identified and we demonstrate a mouse model that finally enables human-relevant risk assessments of ingested, bio-persistent (nano)particles.
PMID:40615368 | DOI:10.1038/s41467-025-60248-9
Utility of a Modified Penlight-Cover Test for Neurolocalization of Lesions Based on Visual Suppression of Nystagmus in Dogs and Cats With Vestibular Disease
J Vet Intern Med. 2025 Jul-Aug;39(4):e70182. doi: 10.1111/jvim.70182.
ABSTRACT
BACKGROUND: Humans with peripheral vestibular disorders can suppress nystagmus through visual fixation, a capability often compromised in those with central vestibular disorders. Bedside tests that exploit this difference can aid neurolocalization in humans. These tests remain unexplored in veterinary medicine.
HYPOTHESIS: Removal of visual input will reveal or enhance nystagmus in animals with peripheral vestibular disease, while animals with central vestibular disease would show little change.
ANIMALS: Twenty-one dogs and cats with peripheral vestibular lesions and 16 with central vestibular lesions. Diagnosis was confirmed by MRI.
METHODS: A prospective study was conducted using a modified penlight-cover test. Because animals cannot be easily instructed to fixate on a visual target, removal of visual input was used as a substitute for eliminating visual fixation, based on the assumption that visual fixation also occurs spontaneously. A 0.5-W LED penlight was shined into one eye while covering the other to eliminate visual input. Nystagmus beat frequency (BF) and subjective evaluation of slow phase velocity (SPV) were recorded before and during penlight application.
RESULTS: In animals with peripheral lesions, BF increased in 33% and SPV in 24% of cases after removal of visual input. Among those with central lesions, only one of 16 showed an increase in BF, and none exhibited an increase in SPV.
CONCLUSIONS: When used alongside the neurological examination, the modified penlight-cover test, could raise suspicion of a peripheral vestibular lesion if it reveals increased BF or SPV.
PMID:40577055 | DOI:10.1111/jvim.70182
Tubular retractors in neuro-oncological surgery: a systematic review and meta-analysis
Neurosurg Rev. 2025 Jun 27;48(1):530. doi: 10.1007/s10143-025-03677-w.
ABSTRACT
Neuro-oncological surgery necessitates a careful balance between maximising tumour resection whilst minimising damage to healthy brain parenchyma. Tubular retractors represent an emerging tool proposed to facilitate in the optimisation of this onco-functional balance. The objective was to evaluate the evidence regarding tubular retractors in neuro-oncological surgery. A systematic review and meta-analysis was performed. Studies reporting on surgical outcomes of tubular retractors in adult neuro-oncological cases were eligible. Medline, Embase, Cochrane Library, ClinicalTrials.gov, and ICTRP were searched to 14th July 2024. Duplicate title/abstract screening, data extraction, and risk of bias assessments were conducted. Prevalence of gross total resection (GTR) and complications were calculated using random effects models. 49 studies were included in the final analysis with a total of 684 patients. Combined pooled prevalence for GTR was 76% (95% CI: 67-85%), whilst for complications was 14% (95% CI: 8-20%). GTR rate by tumour histology was: 52% for gliomas (95% CI: 41-62%), 80% for metastases (95% CI: 65-92%), and 100% for colloid cysts (95% CI: 99-100%). Complication rate by tumour histology was: 16% for gliomas (95% CI: 5-30%), 12% for metastases (95% CI: 1-28%), and 16% for colloid cysts (95% CI: 8-24%). There was no significant difference between tubular retractor brands and GTR or complication rate (p > 0.05). Despite the mounting interest regarding the utility of tubular retractors in neuro-oncological surgery, the current evidence remains largely in the form of case series. Prospective studies with greater sample sizes, longer follow-up, and direct comparison to conventional retraction are now needed.
PMID:40576849 | DOI:10.1007/s10143-025-03677-w