{kind=link}
Epithelial damage and ageing: the perfect storm
Thorax. 2025 May 27:thorax-2024-222060. doi: 10.1136/thorax-2024-222060. Online ahead of print.
ABSTRACT
BACKGROUND: Idiopathic pulmonary fibrosis (IPF) is a progressive disease of lung parenchymal scarring that is triggered by repeated microinjury to a vulnerable alveolar epithelium. It is increasingly recognised that cellular ageing, whether physiological or accelerated due to telomere dysfunction, renders the epithelium less able to cope with injury and triggers changes in epithelial behaviour that ultimately lead to the development of disease.
AIMS: This review aims to highlight how, with increasing age, the alveolar epithelium becomes vulnerable to exogenous insults. We discuss the downstream consequences of alveolar epithelial dysfunction on epithelial phenotype, alveolar repair and pro-pathogenic interactions with other alveolar niche-resident cell types which drive IPF pathogenesis.
NARRATIVE: We highlight how a wide array of cellular mechanisms that maintain cellular homeostasis become dysfunctional with ageing. Waning replicative capacity, genomic stability, mitochondrial function, proteostasis and metabolic function all contribute to a phenotype of vulnerability to 'second hits'. We discuss how in IPF the alveolar epithelium becomes dysfunctional, highlighting changes in repair capacity and fundamental cellular phenotype and how interactions between abnormal epithelium and other alveolar niche-resident cell types perpetuate disease.
CONCLUSIONS: The ageing epithelium is a vulnerable epithelium which, with the cumulative effects of environmental exposures, fundamentally changes its behaviour towards stalled differentiation, failed repair and profibrotic signalling. Further dissection of aberrant epithelial behaviour, and its impact on other alveolar cell types, will allow identification of novel therapeutic targets aimed at earlier pathogenic events.
PMID:40425299 | DOI:10.1136/thorax-2024-222060
Opportunities and challenges for monitoring terrestrial biodiversity in the robotics age
Nat Ecol Evol. 2025 May 22. doi: 10.1038/s41559-025-02704-9. Online ahead of print.
ABSTRACT
With biodiversity loss escalating globally, a step change is needed in our capacity to accurately monitor species populations across ecosystems. Robotic and autonomous systems (RAS) offer technological solutions that may substantially advance terrestrial biodiversity monitoring, but this potential is yet to be considered systematically. We used a modified Delphi technique to synthesize knowledge from 98 biodiversity experts and 31 RAS experts, who identified the major methodological barriers that currently hinder monitoring, and explored the opportunities and challenges that RAS offer in overcoming these barriers. Biodiversity experts identified four barrier categories: site access, species and individual identification, data handling and storage, and power and network availability. Robotics experts highlighted technologies that could overcome these barriers and identified the developments needed to facilitate RAS-based autonomous biodiversity monitoring. Some existing RAS could be optimized relatively easily to survey species but would require development to be suitable for monitoring of more 'difficult' taxa and robust enough to work under uncontrolled conditions within ecosystems. Other nascent technologies (for instance, new sensors and biodegradable robots) need accelerated research. Overall, it was felt that RAS could lead to major progress in monitoring of terrestrial biodiversity by supplementing rather than supplanting existing methods. Transdisciplinarity needs to be fostered between biodiversity and RAS experts so that future ideas and technologies can be codeveloped effectively.
PMID:40404926 | DOI:10.1038/s41559-025-02704-9
Visual preferences for communicating modelling: a global analysis of COVID-19 policy and decision makers
Infect Dis Model. 2025 Apr 23;10(3):924-934. doi: 10.1016/j.idm.2025.04.005. eCollection 2025 Sep.
ABSTRACT
Effective communication of modelling results to policy and decision makers has been a longstanding challenge in times of crises. This communication takes many forms - visualisations, reports, presentations - and requires careful consideration to ensure accurate maintenance of the key scientific messages. Science-to-policy communication is further exacerbated when presenting fundamentally uncertain forms of science such as infectious disease modelling and other types of modelled evidence, something which has been understudied. Here we assess the communication and visualisation of infectious disease modelling results to national COVID-19 policy and decision makers in 13 different countries. We present a synthesis of recommendations on what aspects of visuals, graphs, and plots policymakers found to be most helpful in their COVID-19 response work. This work serves as a first evidence base for developing guidelines on the communication and translation of infectious disease modelling into policy.
PMID:40390802 | PMC:PMC12088752 | DOI:10.1016/j.idm.2025.04.005
Competition between transmission lineages mediated by human mobility shapes seasonal influenza epidemics in the US
Nat Commun. 2025 May 17;16(1):4605. doi: 10.1038/s41467-025-59757-4.
ABSTRACT
Due to its climatic variability, complex mobility networks and geographic expanse, the United States represents a compelling setting to explore the transmission processes that lead to heterogeneous yearly seasonal influenza epidemics. By analyzing genomic and epidemiological data collected in the US from 2014 to 2023, we show that epidemics consisted of multiple co-circulating transmission lineages that could emerge from all regions and often rapidly expanded. Lineage spread was characterized by strong spatiotemporal hierarchies and lineage size correlated with timing of establishment in the US. Mechanistic epidemic simulations, supported by phylogeographic analyses, suggest that competition between lineages on a network of human mobility consistent with commuting flows drove lineage dynamics. Our results suggest that the processes that disseminate viruses nationwide are highly structured, but variability in the short-term processes that determine the locations, timing, and explosiveness of initial epidemic sparks limits predictability of regional and national epidemics.
PMID:40382319 | DOI:10.1038/s41467-025-59757-4
Development of a Novel Epilepsy and Dyskinesia Survey for Large-Scale Characterization of Seizure Semiology in Dogs
J Vet Intern Med. 2025 May-Jun;39(3):e70077. doi: 10.1111/jvim.70077.
ABSTRACT
BACKGROUND: Diagnosing epilepsy and dyskinesia in dogs relies on seizure semiology, laboratory workup, brain imaging, and electroencephalography. Variability in existing epilepsy surveys complicates comparison and impedes epidemiologic and genetic research.
OBJECTIVE: To characterize the semiology of epileptic seizures and dyskinesia episodes using a novel, owner-completed, multi-language online questionnaire.
ANIMALS: A cohort of 606 dogs from 96 breeds with paroxysmal events, perceived by their owners as epilepsy or dyskinesia.
MATERIALS AND METHODS: A comprehensive epilepsy and dyskinesia questionnaire featuring pragmatic seizure categories and video upload was developed in German, Finnish, and English. The reliability of the questionnaire was assessed, and the study cohort analyzed.
RESULTS: The questionnaire demonstrated strong internal consistency and interrater agreement. Owners correctly classified paroxysmal events in 90.1% of cases (95% CI 88.18-92.11). Video footage was submitted from 23.8% (143/606) and supported the seizure type in the questionnaire in 96.5%. The age of onset ranged from 6 months to 6 years in 80.2% (median 2 years; IQR 1-5 years). Generalized (epileptic) convulsive seizures occurred in 58.6% of dogs, non-generalized paroxysmal motor events without convulsions in 58.1%, sudden falls without movement in 6.1%, episodes of impaired awareness in 15.8%, and other unclassified events in 7.1%. Multiple seizure types were reported in 25.2% of the dogs. Labrador Retrievers exhibited a higher prevalence of non-generalized motor events compared to Border Collies, Siberian Huskies, and other breeds (p < 0.001).
CONCLUSIONS: The questionnaire reliably characterizes epileptic seizures and dyskinesia episodes in dogs, making it a valuable tool for large-scale epidemiological and genetic studies.
PMID:40375574 | DOI:10.1111/jvim.70077
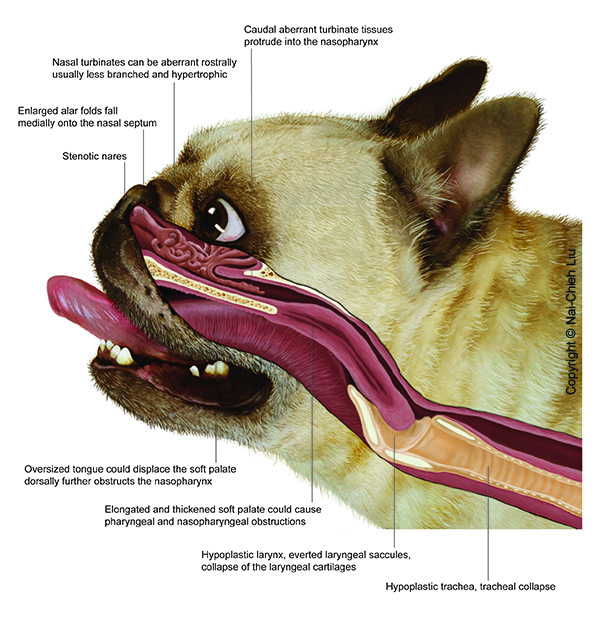
Static respiratory compliance in anaesthetised and intubated brachycephalic dogs with and without brachycephalic obstructive airway syndrome
Vet J. 2025 May 13:106372. doi: 10.1016/j.tvjl.2025.106372. Online ahead of print.
ABSTRACT
The impact of brachycephalic obstructive airway syndrome in dogs (BOAS) on respiratory mechanics is unclear and may affect the choice of ventilation strategies during anaesthesia. This prospective study included 56 client-owned brachycephalic dogs, allocated to be BOAS (n = 26) or non-BOAS dogs (n = 30) based on functional grading. All dogs were anaesthetised using a standardised anaesthetic protocol. Pressure-controlled ventilation was initiated around 30minutes post-induction, maintaining peak inspiratory pressure at 7-12cm H2O. Static respiratory compliance (Cstat) was recorded at predetermined time points in sternal, right and left lateral recumbency. Thorax dimensions were assessed with a tape measure. Body surface area (BSA) was calculated and the ratio Cstat/BSA used as the main outcome variable. Comparison of means/medians, analysis of proportions, the Spearman correlation coefficient and both logistic and linear regression were used for data analysis. P < 0.05 was considered statistically significant. Non-BOAS dogs showed significantly higher Cstat/BSA compared to BOAS dogs in sternal (41.6 (31.1-51.8) vs. 32.9 (24.4 - 39.2), respectively, P = 0.028), right lateral (36.2 (25.7 - 46.4) vs. 27.0 (22.7 - 35.6); P = 0.026) and left lateral (33.6 (22.6 - 45.5) vs. 24.6 (18.4 - 32.2); P = 0.020) recumbencies. For all dogs, the Cstat/BSA ratio was higher in sternal compared to lateral recumbencies. BOAS dogs had a significantly shorter distance between thoracic inlet and last rib compared to non-BOAS dogs (20 ± 4 vs. 23 ± 6cm, respectively; P = 0.043). Reduced respiratory compliance in BOAS-affected dogs should be considered during mechanical ventilation.
PMID:40374099 | DOI:10.1016/j.tvjl.2025.106372
Kinetic and Kinematic Gait Analyses of Dogs with Undersized Cementless Stems (Biphasic Calcium Phosphate Coated) versus Cemented in Total Hip Replacement
Vet Comp Orthop Traumatol. 2025 May 8. doi: 10.1055/a-2591-7747. Online ahead of print.
ABSTRACT
To use kinetic and kinematic analysis to determine whether a cementless femoral implanted with a bioactive coating can be an effective alternative to a cemented femoral stem.In the Cemented group, six dogs were implanted with a 316L stainless steel hip prosthesis. The six dogs in the Cementless group were implanted with a 316L stainless steel hip prosthesis with a biphasic calcium phosphate coating. Kinetic gait analysis was performed before the surgery and at 2, 4, 6, 8, and 12 weeks postoperatively. Kinematic analyses were carried out before the surgery and at 2, 4, 6, 8, 12, and 16 weeks.A slow and sustained improvement in kinetic parameters occurred over time. Dogs implanted with the cemented prosthesis recovered normal preoperative values for maximal hip extension angle by 4 weeks after surgery. Dogs with cementless prosthesis had not recovered normal hip extension by 4 weeks. Despite this short-term change in hip mobility, there were no significant differences in gait between the two groups over the 4-month study period.No differences in gait between cementless and cemented prosthesis were observed throughout the postoperative period to the fourth month. Additionally, compensation and adaptation with gradual recovery of kinetic and kinematic parameters were evident.
PMID:40341514 | DOI:10.1055/a-2591-7747
Enhanced variant neutralisation through glycan masking of SARS-CoV-2 XBB1.5 RBD
Emerg Microbes Infect. 2025 May 6:2502011. doi: 10.1080/22221751.2025.2502011. Online ahead of print.
NO ABSTRACT
PMID:40326334 | DOI:10.1080/22221751.2025.2502011
Emergence and spread of ST5 methicillin-resistant Staphylococcus aureus with accessory gene regulator dysfunction: genomic insights and antibiotic resistance
Microbiol Res. 2025 Apr 25;297:128196. doi: 10.1016/j.micres.2025.128196. Online ahead of print.
ABSTRACT
The globally disseminated Staphylococcus aureus ST5 clone poses a major public health threat due to its multidrug resistance and virulence. Here, we identified an agr-dysfunctional (agrA-I238K) ST5 MRSA clone that has spread across East and Southeast Asia, with recent increases in China since its emergence in the 1970s. Comparative genomic analyses identified distinct single-nucleotide polymorphisms and mobile genetic elements linked to enhanced resistance and virulence. This clone exhibits resistance to seven antimicrobial classes, including third-generation tetracyclines and fusidic acid, and shares phenotypic and genetic similarities with the vancomycin-intermediate S. aureus Mu50 strain, including reduced susceptibility to vancomycin, teicoplanin, and daptomycin. The agrA-I238K mutation attenuates hemolytic activity, increases biofilm formation, and reduces daptomycin susceptibility, suggesting a key role in the clone's success. Our results demonstrate the important role of agrA-I238K mutation in the widespread distribution of agr-dysfunctional MRSA and highlight the importance of genomic surveillance in tracking the spread of agr-dysfunctional ST5 MRSA.
PMID:40311457 | DOI:10.1016/j.micres.2025.128196
Adoption of Biosecurity Practices in Smallholder Dairy Farms in Ethiopia
Transbound Emerg Dis. 2023 Aug 14;2023:2277409. doi: 10.1155/2023/2277409. eCollection 2023.
ABSTRACT
Dairy production is an important livelihood source for smallholder dairy farmers who produce the majority of milk consumed and traded in Ethiopia. Dairy production is, however, constrained by livestock diseases that impact farm productivity, food safety, and animal welfare. Biosecurity measures (BSM) include all risk reduction strategies designed to avoid the introduction of pathogenic infections from outside and minimise the spread of diseases within dairy herds. This study used a cross-sectional survey to investigate the adoption of BSM in dairy farms in Addis Ababa and Oromia regions of Ethiopia. Using a questionnaire, scores for adopted external and internal BSM were calculated based on the Ghent's University Biocheck tool to compare the performance of different farms in Ethiopia. The weighted external biosecurity score was 49.1%, which was below average (below 50% adoption), while the weighted internal biosecurity score was 55.5%. Low adoption of crucial BSM increases the risk of disease introduction into dairy farms and transmission within herds. Adoption of BSM at the farm level was driven by individual, demographic, and socio-economic drivers, including education, farming system, milk value chain, and farming experience among others. Results of this research reveal low adoption of BSM and the imperative to encourage farmers to implement BSM can lead to a reduction in disease pressures and, thus, a reduction in antibiotic use and increased dairy farms productivity, and improved animal health and welfare. Farmers can be encouraged through proactive engagement with veterinarians and extension professionals. Moreover, creating a favourable policy environment can support farmers to adopt and implement BSM, given the known fact that "prevention is better and cheaper than curing diseases."
PMID:40303828 | PMC:PMC12016702 | DOI:10.1155/2023/2277409
Case Series of Canine Myasthenia Gravis: A Classification Approach With Consideration of Seronegative Dogs
J Vet Intern Med. 2025 May-Jun;39(3):e70113. doi: 10.1111/jvim.70113.
ABSTRACT
BACKGROUND: Myasthenia gravis (MG) is categorized into several subgroups, including seronegative MG. Seronegative human patients are well documented, but seronegative dogs remain clinically uncharacterized and their prevalence unknown.
OBJECTIVES: This study aims to evaluate the clinical presentation, diagnosis, treatment, and outcome of canine MG subgroups.
ANIMALS: One hundred sixty-seven owner-owned dogs diagnosed with MG from three referral centers.
METHODS: Retrospective case series. We classified myasthenic dogs into subgroups, adhering to human guidelines.
RESULTS: We classified 167 dogs into four subgroups: acetylcholine receptor (AChR) antibody-positive generalized (49.7%, n = 83/167), focal (19.2%, n = 32/167) and thymoma-associated MG (9%, n = 15/167) and seronegative MG (22.2%, n = 37/167). Dogs with thymoma-associated MG were older (median 102 months; Interquartile Range (IQR) 96-120; p < 0.001) and seronegative dogs were younger (median 30 months; IQR 11.5-66; p = 0.017), compared to the generalized subgroup (median 67 months; IQR 36-96). Seronegative dogs presented less frequently with megaesophagus, compared to the generalized subgroup (63.8% vs. 85.7%; Odds Ratio 3.4; 95% confidence intervals (C.I.) 1.4-8.9; p = 0.025). Myasthenic dogs' survival time was significantly reduced when thymoma (Hazard Ratio (H.R.) 3.7; 95% C.I. 1.4-9.9; p = 0.028) or esophageal weakness (H.R. 3.8; 95% C.I. 2.0-7.0; p < 0.001) was present. Conversely, a higher likelihood of remission was achieved when esophageal weakness was absent (H.R. 3.8; 95% C.I. 1.4-10.0; p = 0.007).
CONCLUSION AND CLINICAL IMPORTANCE: Dogs with seronegative MG are more common than previously reported. Myasthenic subgroups differ in presentation and outcome, with esophageal weakness key to survival and remission. Diagnostic tests for seronegative dogs and effective treatments for esophageal weakness in myasthenic dogs are urgently needed.
PMID:40298067 | DOI:10.1111/jvim.70113
Antibiotic-Loaded Polymer-Calcium Phosphate Scaffold for Treating Orthopedic Device-Related Infection in a Rabbit Segmental Bone Defect Model
J Biomed Mater Res A. 2025 May;113(5):e37917. doi: 10.1002/jbm.a.37917.
ABSTRACT
Treatment of orthopedic device-related infection (ORDI) generally requires a combination of medical and surgical interventions for successful treatment outcomes. Many cases are treated with a two-stage revision, whereby contaminated implants and necrotic tissues are removed, and dead space is managed with a temporary, non-resorbable polymethyl methacrylate (PMMA) spacer loaded with antibiotics. Weeks later, this is replaced with a bone graft or similar material to aid bone healing. However, this two-stage approach is quite a burden for the patient, and infection may still recur. The use of a 3D-printed, absorbable, antibiotic-releasing material that also promotes bone healing would be a promising alternative that produces the exact geometry of the missing bone and eliminates the need for a second surgery. In this study, we investigated whether a novel 3D-printed, antibiotic-loaded, osteoconductive calcium phosphate scaffold (CPS) is effective in the single-stage revision of an infected segmental bone defect model in rabbits. A 5-mm segmental defect of the radius of female New Zealand White rabbits (n = 64), stabilized with cerclage wire, was inoculated with Staphylococcus aureus. After 4 weeks, the infected bone fragment was removed, the site debrided, and the bone defect was either left empty (Control group) or filled with a PMMA spacer with gentamicin, CPS loaded with rifampicin or non-loaded CPS. The animals were also managed with systemic cefazolin for 4 weeks. An additional group received vancomycin-loaded CPS without adjunctive systemic antibiotic therapy. All animals were euthanized 8 weeks after revision and assessed by quantitative bacteriology or semiquantitative histopathology. The antibiotic-loaded scaffolds (PMMA-Genta and CPS-Rif) in the animals receiving systemic antibiotic treatment resulted in a reduction in bacterial count at euthanasia compared to controls (rabbits receiving systemic antibiotic alone and in which the defect was left empty). The PMMA-Genta induced a significant CFU reduction (p = 0.0486) compared to controls. The infection rate was also reduced from 80% in the control group to 50% for the groups receiving local and systemic antibiotics. The CPS-Vanco group for local delivery without adjunctive systemic antibiotic therapy resulted in a lower infection rate, but the CFUs in these samples at euthanasia were comparable with those of the control group. The findings show that treating an ODRI with PMMA-Genta yields the best results for infection eradication; however, it does not provide the reconstruction opportunity that the antibiotic-loaded CPS does. Even though it is not comparable to the PMMA-Genta, the antibiotic-loaded CPS showed a reduction in infection rates. The use of local antibiotics alone is insufficient to eradicate the infection.
PMID:40296342 | DOI:10.1002/jbm.a.37917
Horizontal transfer of nuclear DNA in transmissible cancer
Proc Natl Acad Sci U S A. 2025 May 6;122(18):e2424634122. doi: 10.1073/pnas.2424634122. Epub 2025 Apr 22.
ABSTRACT
Horizontal transfer of nuclear DNA between cells of host and cancer is a potential source of adaptive variation in cancer cells. An understanding of the frequency and significance of this process in naturally occurring tumors is, however, lacking. We screened for this phenomenon in the transmissible cancers of dogs and Tasmanian devils and found an instance in the canine transmissible venereal tumor (CTVT). This involved introduction of a 15-megabase dicentric genetic element, composed of 11 fragments of six chromosomes, to a CTVT sublineage occurring in Asia around 2,000 y ago. The element forms the short arm of a small submetacentric chromosome and derives from a dog with ancestry associated with the ancient Middle East. The introduced DNA fragment is transcriptionally active and has adopted the expression profile of CTVT. Its features suggest that it may derive from an engulfed apoptotic body. Our findings indicate that nuclear horizontal gene transfer, although likely a rare event in tumor evolution, provides a viable mechanism for the acquisition of genetic material in naturally occurring cancer genomes.
PMID:40261943 | DOI:10.1073/pnas.2424634122
Capsular immunity is necessary for protection against some but not all strains of Glaesserella parasuis
Vet Microbiol. 2025 Apr 11;305:110509. doi: 10.1016/j.vetmic.2025.110509. Online ahead of print.
ABSTRACT
Glaesserella parasuis is the causative agent of Glässer's disease in pigs and results in significant losses to the swine industry annually. Due to the serovar and strain specific response associated with many bacterin vaccines, there has been difficulty generating broad heterologous protection. Here, an unencapsulated G. parasuis mutant (HS069∆cap) was assessed as a bacterin vaccine and compared to a bacterin made from the encapsulated parent strain, against challenge with the homologous, parent strain (serovar 5) as well as four heterologous challenge strains (serovar 1, 4, 5, and 14). Both the HS069 and HS069∆cap bacterins generated high titers to the homologous and heterologous strains. The HS069∆cap bacterin was able to provide protection against the parent strain as well as 12939 (serovar 1), 2170B (serovar 4), and MN-H (serovar 13), but was unable to protect animals from challenge with Nagasaki (serovar 5). In contrast, the HS069 bacterin was able to provide protection against all challenge strains, showing the importance of serovar specific immunity against the challenge strain Nagasaki. This appears to be due to the production of a more abundant and well-organized capsule in Nagasaki as compared to HS069. This study indicates HS069∆cap is a good candidate strain for bacterin development; however, it may be less able to provide protection against highly encapsulated strains of G. parasuis.
PMID:40250105 | DOI:10.1016/j.vetmic.2025.110509
Similarity of drug targets to human microbiome metaproteome promotes pharmacological promiscuity
Pharmacogenomics J. 2025 Apr 17;25(3):9. doi: 10.1038/s41397-025-00367-0.
ABSTRACT
Similarity between candidate drug targets and human proteins is commonly assessed to minimize the occurrence of side effects. Although numerous drugs have been found to disrupt the health of the human microbiome, no comprehensive comparison between established drug targets and the human microbiome metaproteome has yet been conducted. Therefore, herein, sequence and structure alignments between human and pathogen drug targets and representative human gut, oral, and vaginal microbiome metaproteomes were performed. Both human and pathogen drug targets were found to be similar in sequence, function, structure, and drug binding capacity to proteins in diverse pathogenic and non-pathogenic bacteria from all three microbiomes. The gut metaproteome was identified as particularly susceptible overall to off-target effects. Certain symptoms, such as infections and immune disorders, may be more common among drugs that non-selectively target host microbiota. These findings suggest that similarities between human microbiome metaproteomes and drug target candidates should be routinely checked.
PMID:40246834 | DOI:10.1038/s41397-025-00367-0
European guidelines on treatment and supportive measures in chronic neutropenias: A consensus between the European Hematology Association and the EuNet-INNOCHRON COST Action based on a systematic evidence review
Hemasphere. 2025 Apr 16;9(4):e70113. doi: 10.1002/hem3.70113. eCollection 2025 Apr.
ABSTRACT
The treatment of chronic neutropenias and control of neutropenia-related infections remain challenging topics for pediatric and adult hematologists. This article aims to fill the gap in the treatment of neutropenias and, in combination with the previously published European guidelines on diagnosis of neutropenias, gives complete and comprehensive guidance on the whole management of patients with neutropenia. In terms of methodology, an Evidence-Based Medicine team produced an evidence synthesis of the literature on the treatment of neutropenias. Then, according to the robustness of the evidence, consensus recommendations were elaborated and voted by an expert's panel from the Cooperation in Science and Technology European Network for the Innovative Diagnosis and Treatment of Chronic Neutropenias (https://eunet-innochron.eu/) and the Specialized Working Group on Granulocytes and Constitutional Bone Marrow Failure Syndromes of the European Hematology Association. Whenever evidence was not available, recommendations were based on the expert's panel opinion. Consensus-based recommendations are related to granulocyte colony-stimulating factor indications and schedule of administration, indications for hematopoietic stem cell transplantation, supportive treatments and measures, and new treatments that have been evolving over the recent years. These guidelines, rather than a numerical correction of the absolute neutrophil count, suggest a holistic, patient-centered approach aiming at optimizing the management of chronic neutropenic patients and offering valuable and practical guidance to the hematologists for their daily clinical practice.
PMID:40242664 | PMC:PMC12001981 | DOI:10.1002/hem3.70113
What's so special about special issues: Highlighting a central role of <em>parasitology</em> to support specific innovations and advance progress within our discipline
Parasitology. 2025 Apr 14:1-5. doi: 10.1017/S0031182025000125. Online ahead of print.
NO ABSTRACT
PMID:40223707 | DOI:10.1017/S0031182025000125
Astrocytic RNA editing regulates the host immune response to alpha-synuclein
Sci Adv. 2025 Apr 11;11(15):eadp8504. doi: 10.1126/sciadv.adp8504. Epub 2025 Apr 11.
ABSTRACT
RNA editing is a posttranscriptional mechanism that targets changes in RNA transcripts to modulate innate immune responses. We report the role of astrocyte-specific, ADAR1-mediated RNA editing in neuroinflammation in Parkinson's disease (PD). We generated human induced pluripotent stem cell-derived astrocytes, neurons and cocultures and exposed them to small soluble alpha-synuclein aggregates. Oligomeric alpha-synuclein triggered an inflammatory glial state associated with Toll-like receptor activation, viral responses, and cytokine secretion. This reactive state resulted in loss of neurosupportive functions and the induction of neuronal toxicity. Notably, interferon response pathways were activated leading to up-regulation and isoform switching of the RNA deaminase enzyme, ADAR1. ADAR1 mediates A-to-I RNA editing, and increases in RNA editing were observed in inflammatory pathways in cells, as well as in postmortem human PD brain. Aberrant, or dysregulated, ADAR1 responses and RNA editing may lead to sustained inflammatory reactive states in astrocytes triggered by alpha-synuclein aggregation, and this may drive the neuroinflammatory cascade in Parkinson's.
PMID:40215316 | DOI:10.1126/sciadv.adp8504
Modelling Human Gut-Microbiome Interactions in a 3D Bioelectronic Platform
Small Sci. 2024 Apr 22;4(6):2300349. doi: 10.1002/smsc.202300349. eCollection 2024 Jun.
ABSTRACT
The role of the gut microbiome in various aspects of health and disease is now a well-established concept in modern biomedicine. Numerous studies have revealed links between host health and microbial activity, spanning from digestion and metabolism to autoimmune disorders, stress and neuroinflammation. However, the exact mechanisms underlying this complex cross-talk still remain a mystery. Conventionally, studies examining host-microbiome interactions rely on animal models, but translation of such findings into human systems is challenging. Bioengineered models represent a highly promisingapproach for tackling such challenges. Here, a bioelectronic platform, the e-transmembrane, is used to establish a 3D model of human intestine, to study the effects of microbiota on gut barrier integrity. More specifically, how postbiotics and live bacteria impact the morphology and function of the intestinal barrier is evaluated. e-Transmembrane devices provide a means for in-line and label-free continuous monitoring of host-microbe cross-talk using electrochemical impedance spectroscopy, revealing distinct patterns that emerge over 24 hours. Microscopy and quantification of molecular biomarkers further validate the differential effects of each bacterial intervention on the host tissue. In addition, a framework to better study and screen drug candidates and potential therapeutic/dietary interventions, such as postbiotics and probiotics, in more physiologically relevant human models is provided.
PMID:40212761 | PMC:PMC11935216 | DOI:10.1002/smsc.202300349
Common variants in the CPT1A gene are associated with cataracts in Northern breeds of domestic dog
PLoS One. 2025 Apr 4;20(4):e0320878. doi: 10.1371/journal.pone.0320878. eCollection 2025.
ABSTRACT
Primary hereditary cataract affects many purebred domestic dog breeds and is a major cause of visual impairment in dogs. Cataracts are common in Northern breeds such as the Siberian Husky, Alaskan Malamute and Samoyed, but their aetiology is currently unknown. Only two genetic loci are known to be causally related to primary hereditary cataracts in the dog. To search for genetic loci associated with cataracts in Northern breeds, we used a genome-wide association study approach in three breeds-Siberian Husky, Alaskan Malamute and Samoyed. Cases were defined as dogs with bilateral posterior polar subcapsular cataracts and controls were at least four years of age with no evidence of cataracts or other ocular abnormality. We found a genome-wide statistical association for cataracts in the Siberian Husky on canine chromosome 18 (P-value: 1.1 x 10 - 7), which was independently replicated in a second larger case-control set (P-value 9.8 x 10 - 29). The Samoyed breed also showed evidence for association in the same genomic region (P-value: 2.4 x 10 - 5). We subsequently used targeted resequencing of the associated region (6.5 Mb) in ten Siberian Huskies and whole genome sequencing of a Husky, Malamute, Samoyed and Norwegian Buhund case to conduct fine-mapping and screen for candidate causal variants. These analyses identified a region of linkage disequilibrium in the four breeds containing common variants in the carnitine palmitoyltransferase 1A (CPT1A) gene that are strongly associated with bilateral posterior polar subcapsular cataracts in the Siberian Husky, Samoyed, Icelandic Sheepdog and Norwegian Buhund and we demonstrate that CPT1A is expressed in the dog lens and retina through RNAseq. Our findings represent a novel locus for cataracts in dogs. However, further work is needed to elucidate the pathophysiology underlying the association between CPT1A and cataracts in Northern breeds.
PMID:40184359 | DOI:10.1371/journal.pone.0320878