{kind=link}
Characterisation of <em>Ornithobacterium hominis</em> colonisation dynamics and interaction with the nasopharyngeal microbiome in a South African birth cohort
bioRxiv [Preprint]. 2025 May 24:2025.05.24.655922. doi: 10.1101/2025.05.24.655922.
ABSTRACT
Ornithobacterium hominis is a recently described Gram-negative bacterium that colonises the human nasopharynx and may be associated with poor upper respiratory tract health. Here, we describe the isolation of O. hominis from samples collected from a South African birth cohort, creating the first archive of cultured strains of the species from Africa. Sequenced genomes from this archive reveal that South African O. hominis is more similar to Australian strains than those from Southeast Asia, and that it may share genes with other members of the microbiome that are relevant for virulence, colonisation, and antibiotic resistance. Leveraging existing microbiome data from the cohort, O. hominis was found to be closely associated with bacterial co-colonisers that are rare in non-carrier individuals, including Suttonella, Helcococcus, Moraxella spp., and Gracilibacteria. Their collective acquisition has a significant impact on the diversity of nasopharyngeal communities that contain O. hominis. Individuals who have not yet acquired O. hominis have a higher abundance of Moraxella (particularly M. lincolnii) than individuals who never acquire O. hominis, suggesting that this could be a precursor state for successful colonisation. Finally, a novel co-coloniser species, Helcococcus ekapensis, was successfully isolated and sequenced.
PMID:40475515 | PMC:PMC12139837 | DOI:10.1101/2025.05.24.655922
How does policy modelling work in practice? A global analysis on the use of epidemiological modelling in health crises
PLOS Glob Public Health. 2025 Jun 6;5(6):e0004675. doi: 10.1371/journal.pgph.0004675. eCollection 2025.
ABSTRACT
This study examines the use and translation of epidemiological modelling by policy and decision makers in response to the COVID-19 outbreak. Prior to COVID-19, there was little readiness for global health systems, and many science-policy networks were assembled ad-hoc. Moreover, in the field of epidemiological modelling, one with significant sudden influence, there is still no international guidance or standard of practice on how modelled evidence should guide policy during major health crises. Here we use a multi-country case study on the use of epidemiological modelling in emergency COVID-19 response, to examine the effective integration of crisis science and policy in different countries. We investigated COVID-19 modelling-policy systems and practices in 13 countries, spanning all six UN geographic regions. Data collection took the form of expert interviews with a range of national policy/ decision makers, scientific advisors, and modellers. We examined the current use of epidemiological modelling, introduced a classification framework for outbreak modelling and policy on which best practice can be structured, and provided preliminary recommendations for future practice. Full analysis and interpretation of the breadth of interview responses is presented, providing evidence for the current and future use of modelling in disease outbreaks. We found that interviewees in countries with a similar size and type of modelling infrastructure, and similar level of government interaction with modelling reported similar experiences and recommendations on using modelling in outbreak response. From this, we introduced a helpful grouping of country experience upon which a tailored future best practice could be structured. We concluded the article by outlining context-specific activities that modellers and policy actors could consider implementing in their own countries. This article serves as a first evidence base for the current use of modelling in a recent major health crisis and provides a robust framework for developing epidemiological modelling-to-policy best practice.
PMID:40478854 | DOI:10.1371/journal.pgph.0004675
The Value of Neutrophil Cell Population Data Parameters as Markers of Systemic Inflammation in Dogs and Cats
Vet Clin Pathol. 2025 Jun 6. doi: 10.1111/vcp.70029. Online ahead of print.
ABSTRACT
BACKGROUND: Neutrophil cell population data (CPD), including fluorescent light intensity (NE-SFL) and side scatter (NE-SSC), are promising inflammatory markers in human sepsis but remain unexplored in dogs and cats.
OBJECTIVES: Determine the diagnostic utility of NE-SSC and NE-SFL for detecting systemic inflammation in dogs and cats.
METHODS: Dogs and cats with archived CPD, blood films, and acute phase protein (APP) measurements were included. Increased C-reactive protein (CRP) in dogs and Serum Amyloid A (SAA) in cats were considered indicative of systemic inflammation. CPD was compared with APPs, white cell count (WCC), neutrophil count, band neutrophil count, and toxic change grade. Optimal cut-offs and associated sensitivities and specificities were calculated using ROC curve analysis. Correlations were assessed using Spearman's coefficient.
RESULTS: NE-SFL and NE-SSC were significantly increased in dogs and cats with systemic inflammation. The area under the curve (AUC) of NE-SFL was higher than that of NE-SSC, WCC, and band neutrophil count in both dogs (0.82) and cats (0.77). The optimal NE-SFL cut-off for detecting systemic inflammation was > 41.7 ch in dogs (sensitivity 80%; specificity 66%) and > 37.4 ch in cats (sensitivity 75%; specificity 67%). NE-SFL was positively correlated with APPs, WCC, neutrophil count, and band neutrophil count in both species. NE-SSC was positively correlated with APPs in both species and, in dogs, also with WCC, neutrophil count, and band neutrophil count.
CONCLUSION: CPD, particularly NE-SFL, is a promising marker of inflammation in dogs and cats and could be especially useful when APP quantification or blood smear examination are unavailable.
PMID:40476643 | DOI:10.1111/vcp.70029
Computationally designed haemagglutinin with nanocage plug-and-display elicits pan-H5 influenza vaccine responses
Emerg Microbes Infect. 2025 Jun 6:2511132. doi: 10.1080/22221751.2025.2511132. Online ahead of print.
ABSTRACT
The increasing spread of highly pathogenic avian influenza (HPAI) A/H5 viruses poses a pandemic threat. Circulating clade 2.3.4.4b viruses have demonstrated rapid transcontinental dissemination, extensive reassortment, epizootic spread and potential sustained mammal-to-mammal transmission, signifying a heightened risk of becoming a human pathogen of high consequence. A broadly protective, future-proof vaccine against multiple clades of H5 influenza is urgently needed for pandemic preparedness. Here, we combine two novel vaccine technologies to generate a Digitally Immune Optimised and Selected H5 antigen (DIOSvax-H5inter) displayed multivalently on the mi3 nanocage using the SpyTag003/SpyCatcher003 conjugation system. Mice immunised with DIOSvax-H5inter Homotypic Nanocages at low doses demonstrate potent, cross-clade neutralising antibody and T cell responses against diverse H5 strains. DIOSvax-H5inter Homotypic Nanocages provide a scalable vaccine candidate with the potential for pan-H5 protection against drifted or newly emergent H5 strains. This World Health Organization preferred characteristic is essential for prospective strategic stockpiling in the pre-pandemic phase.Trial registration: ClinicalTrials.gov identifier: NCT06145178..
PMID:40476519 | DOI:10.1080/22221751.2025.2511132
Comparative performance and age dependence of tuberculin and defined antigen bovine tuberculosis skin tests assessed with Bayesian latent class analysis
Sci Rep. 2025 Jun 5;15(1):19728. doi: 10.1038/s41598-025-05223-6.
ABSTRACT
Tuberculin skin tests (TST), the primary diagnostic tool for bovine tuberculosis (bTB), cross-react with BCG vaccine. Recently developed defined antigen skin tests (DSTs) aim to differentiate infected amongst vaccinated animals. We evaluated the field performance of different interpretations of the TST and DSTs relative to IGRA and IDEXX M. bovis antibody tests. This panel of tests was assessed in 446 unvaccinated cattle across 22 Ethiopian dairy herds using Bayesian latent class models. We extended the standard Walter-Hui model to include age-related effects to explore evidence of the presence of diagnostic anergy. The latent class models estimate sensitivity and specificity of the DSTs to be between 84-88% and 79-85% respectively. The DSTs perform intermediately between the comparative intradermal test (CIT, sensitivity 77%, specificity 100%) and single intradermal test (SIT, sensitivity 99%, specificity 76%). We observed significant age-related declines in test sensitivity, most notably for CIT (declining from 75 to 52% over 9 years) and DST10 (83% to 68%), while other tests showed more stable sensitivity across age groups. This variable pattern across tests suggests mechanisms beyond simple age-related anergy. Together, these findings demonstrate that DSTs' superior sensitivity to CIT and comparable or better specificity than SIT, combined with their ability to distinguish vaccinated animals, creates a viable pathway for implementing BCG vaccination programs. Given the absence of any gold standard definition of infection with bTB, latent class analyses are essential to assess the relative performance of different diagnostic tests. While our results provide encouraging news for the sensitivity of the new DST tests, the high prevalence of bTB within our study population makes our design underpowered to assess the specificity of the DSTs. Future research, including assessment of the specificity of DSTs in disease-free populations and optimization of test formulation and validation through large-scale field trials is essential to fully establish the case for use in vaccination and surveillance programs.
PMID:40473835 | DOI:10.1038/s41598-025-05223-6
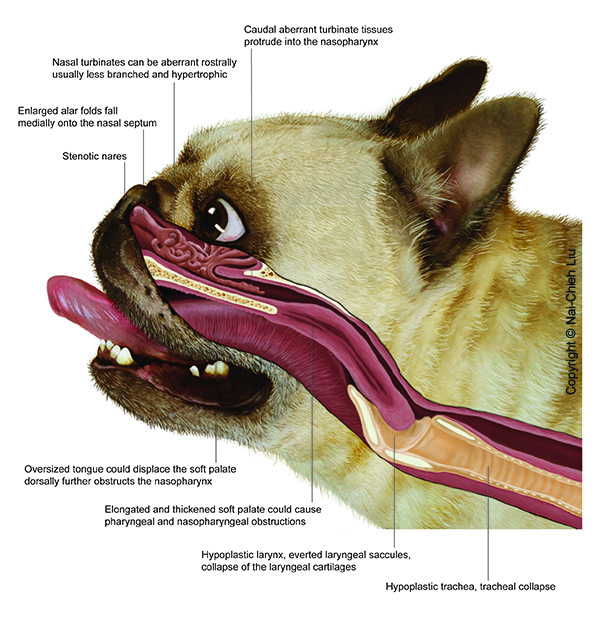
Complication rate and outcomes of laryngeal cuneiformectomy in dogs with advanced laryngeal collapse
Vet Surg. 2025 Jun 2. doi: 10.1111/vsu.14270. Online ahead of print.
ABSTRACT
OBJECTIVE: To describe the complication rate and outcomes of dogs undergoing multilevel airway surgery for brachycephalic airway syndrome (BOAS) with and without the addition of uni- or bilateral cuneiformectomy.
STUDY DESIGN: Retrospective study.
ANIMALS: A total of 180 dogs undergoing BOAS surgery: 94 dogs undergoing modified multilevel surgery (non-PC); 86 additionally undergoing cuneiformectomy (PC).
METHODS: Case records from the University of Cambridge and Animal Health Trust databases between 2014 and 2021 were analyzed including data on laryngeal collapse grade, respiratory functional grading scores, BOAS index, hospitalization length and complications.
RESULTS: Neither the incidence risk of overall (non-PC = 19.4%, PC = 16.3%, p = .758), nor major (non-PC = 7.4%, PC = 11.6%, p = .482) complications differed between non-PC and PC dogs. Median hospitalization duration (non-PC = 1 day, PC = 1 day) did not differ between the two groups (p = .743). Both BOAS grade (median reduction = 1, p < .0001) and BOAS index (median reduction = 28.5%, p < .0001) reduced in dogs that underwent cuneiformectomy. Lower BCS was associated with increased postoperative complications (odds ratio = 0.452, p = .004) when preoperative BOAS grade and gender were controlled.
CONCLUSION: Cuneiformectomy was not associated with a higher incidence risk of complications than multilevel BOAS surgery alone. Significant improvements in respiratory parameters were observed following cuneiformectomy in addition to multilevel airway surgery.
CLINICAL SIGNIFICANCE: Cuneiformectomy represents a safe and effective adjunctive technique to manage higher grade laryngeal collapse in dogs with BOAS.
PMID:40457630 | DOI:10.1111/vsu.14270
Junior Clinical Training Scholar in Small Animal Studies X 6
Six scholarships available to start on Monday 01 December 2025 for 12.5 months.
SCHOLARSHIP AWARD: £21,970.00 (TAX EXEMPT) per annum.
University accommodation package, see details below.
Applications are invited for this one-year post-graduate training programme based in the Queen's Veterinary School Small Animal Hospital. On site accommodation is available for £300 per month including bills.
Junior Clinical Training Scholars will receive training and tuition as they rotate through anaesthesia, cardiology, diagnostic imaging, orthopaedics, dermatology, internal medicine, neurology, oncology, clinical pathology and soft tissue surgery and be supervised by recognised specialists in each field. Scholars will also have responsibility for primary care cases, and be involved in supervision and guidance of final year veterinary students. Scholars will be an integral part of the out of hours care of animals within the hospital, especially within the intensive care unit.
Summary of benefits
- High residency success rate - 74% of our interns have gone on to do a residency and 100% of interns who have been pursuing a residency have successfully achieved a residency or completed a specialist internship programme. We have 50 successful residency applications from intern cohorts 2015-2022 and 2217 diplomates, so far!
- Competitive tax-free stipend including accommodation in Central Cambridge and bills included package
- Truly rotating internship through all specialties including flexibility to pursue extra time in rotations of your choice!
- Good work-life balance with manageable weekend and night work
- University library and journal access
- Monthly seminars with complimentary food and drink!
- 4 weeks of elective/dedicated research time on top of holidays
- Academic opportunities, e.g. teach Cambridge students during rotations and College supervision opportunities; weekly department research and clinical seminars; journal and book clubs
- Proven track-record with publications and research projects with guidance on presentation and scientific writing skills.
- Assigned intern supervisor: - regular progress meetings, interview practice, provision of professional references and CV/cover letter proof reading by experienced senior clinicians to aid residency applications
- Generous CPD allowance and encouragement to present at scientific meetings
- RECOVER CPR training
- First opinion service including surgical cases
- A number of service-specific internships and residency opportunities encourage career progression following internship
Candidates must be Members of the Royal College of Veterinary Surgeons (RCVS) and the following skills and experience are desirable: - Surgical experience in dogs and cats - Completion of 1 year in primary care veterinary practice in the UK - For applicants for whom English is not their first language, a score of 7.5 in IELTS (with no element under 7), or a score of 100 in TOEFL (with no element less than 24).
For further benefits and details on the Internship: https://www.vet.cam.ac.uk/study/cts/jcts1/smallanimal
Informal enquiries should be directed to the Internship Directors, by email: internship.enquiries@vet.cam.ac.uk.
Please note: The ability to take up this Scholarship is contingent upon you being able to evidence your right to work in the UK, or through gaining the right to work via the UK immigration system. Evidence will need to be provided before an offer can be made. Regrettably, this Scholarship is not suitable for sponsorship via the Skilled Worker or Temporary Worker visa routes as the minimum requirements cannot be met.
An application form (JCTS1) and information pack can be downloaded from the link below or via the following website: http://www.vet.cam.ac.uk/job
Applicants should supply a completed Junior Clinical Training Scholarship Application Form (JCTS 1), a CV and Covering Letter giving reasons for wishing to undertake the JCTS in the Department of Veterinary Medicine, University of Cambridge.
Applications should be submitted via e-mail to: vetmed@vet.cam.ac.uk with the above documents as one attachment, by the closing date stated. Please quote reference PP46094 on your application and in any correspondence about this vacancy.
The deadline for applications is midnight on Monday 30 June 2025.
Applications will be monitored regularly, and we may contact candidates prior to the closing date. We reserve the right to close this vacancy early if we receive sufficient applications or extend the closing date if necessary. Therefore, if you are interested, please submit your application as early as possible.
The University actively supports equality, diversity and inclusion and encourages applications from all sections of society.
Junior Clinical Training Scholar in Diagnostic Imaging
Scholarship award: £21,970.00 (Tax Exempt) per annum.
University accommodation package, see details below.
Start date: from 15 July 2025, or as soon as possible thereafter, for 12 months.
Applications are invited from recently qualified veterinarians for this twelve month post-graduate training programme offering high-quality, post-graduate training in Veterinary Diagnostic Imaging. The emphasis will be on gaining practical clinical experience in Veterinary Diagnostic Imaging under the supervision of board-certified diplomates. On site accommodation is available for £300 per month including bills.
Applicants must be a Member of the Royal College of Veterinary Surgeons or hold a veterinary degree qualifying them for membership.
Summary of benefits
- Competitive tax-free stipend including accommodation in Central Cambridge and bills included package
- Good work-life balance with manageable weekend and night work
- University library and journal access
- Academic opportunities, e.g. teach Cambridge students during rotations and College supervision opportunities; weekly department research and clinical seminars; journal and book clubs
- Proven track-record with publications and research projects with guidance on presentation and scientific writing skills.
- Assigned intern supervisor: - regular progress meetings, interview practice, provision of professional references and CV/cover letter proof reading by experienced senior clinicians to aid residency applications
- Generous CPD allowance and encouragement to present at scientific meetings
We would welcome anyone wishing to apply for this scholarship to arrange a visit to the hospital to meet the team and find out more. To arrange a visit to the hospital and for informal enquiries about the scholarship programme, please contact Marie-Aude Genain on mag72@cam.ac.uk.
Please note: The ability to take up this Scholarship is contingent upon you being able to evidence your right to work in the UK, or through gaining the right to work via the UK immigration system. Evidence will need to be provided before an offer can be made. Regrettably, this Scholarship is not suitable for sponsorship via the Skilled Worker or Temporary Worker visa routes as the minimum requirements cannot be met.
An application form (JCTS1) and information pack can be downloaded from the link below or via the following website: http://www.vet.cam.ac.uk/job
Applicants should supply a completed Junior Clinical Training Scholarship Application Form (JCTS 1), a CV and Covering Letter giving reasons for wishing to undertake the JCTS in the Department of Veterinary Medicine, University of Cambridge.
Applications should be submitted via e-mail to: vetmed@vet.cam.ac.uk with the above documents as one attachment, by the closing date stated.
The deadline for applications is midnight on Sunday, 22 June 2025.
Please quote reference PP46093 on your application and in any correspondence about this vacancy.
The University actively supports equality, diversity and inclusion and encourages applications from all sections of society.
Bacteriophage-Antibiotic Synergy Enhances Therapeutic Efficacy Against Multidrug-Resistant Klebsiella Pneumoniae Infections
J Appl Microbiol. 2025 May 29:lxaf131. doi: 10.1093/jambio/lxaf131. Online ahead of print.
ABSTRACT
AIMS: This study aims to evaluate the therapeutic efficacy of bacteriophage therapy alone or in combination with antibiotics in the treatment of acute infection caused by multidrug-resistant (MDR) Klebsiella pneumoniae.
METHODS AND RESULTS: In this study, we isolated and characterized a lytic bacteriophage vB_Kpn_FOPMU1, which exhibits potent antibacterial activity against K. pneumoniae. Whole-genome sequencing identified vB_Kpn_FOPMU1 as a member of the Przondovirus genus and revealed the presence of key lysis-associated genes, including those encoding endolysin, holin, and Rz-like spanin proteins. In vitro work demonstrated that incubation of bacteriophage and cefotaxime with K. pneumoniae significantly decreased the MIC of cefotaxime from 128µg mL-1 to 1 µg mL-1, indicating strong synergistic activity. Using a murine model of acute K. pneumoniae lung infection, we further demonstrated that the combination therapy significantly enhanced bacterial clearance compared to phage monotherapy. This synergistic approach restored sensitivity of K. pneumoniae to cefotaxime, prevented the emergence of phage-resistant bacterial mutants, and achieved superior bacterial eradication from both the lung and blood. Moreover, administration of the phage-antibiotic combination resulted in complete protection of infected mice, with a 100% survival rate, compared to a 60% survival rate observed in animals that received phage monotherapy. Therapeutic application of the bacteriophage-cefotaxime combination resulted in significantly improved lung pathology, characterized by reduced inflammatory cell infiltration and diminished tissue damage, compared to bacteriophage monotherapy.
CONCLUSION: Our findings underscore the potential of bacteriophage-antibiotic synergy as a promising therapeutic strategy to combat MDR K. pneumoniae infections and mitigate the risk of phage resistance development.
PMID:40440204 | DOI:10.1093/jambio/lxaf131
Single-cell transcriptomic analysis of canine insulinoma reveals distinct sub-populations of insulin-expressing cancer cells
Vet Oncol. 2025;2(1):13. doi: 10.1186/s44356-025-00026-3. Epub 2025 May 26.
ABSTRACT
Canine malignant insulinoma is a rare, highly metastatic and life-threatening neuroendocrine tumour of pancreatic beta cells. To map the single-cell transcriptomic landscape of canine insulinoma for the first time, transcriptomic profiles of 5,532 cells were captured from two spontaneous insulinomas (Patient 1 and 2) and one associated metastasis (Patient 2) in two Boxer dogs. Distinct cancer, endocrine, and immune cell populations were identified. Notably, all three tumour samples contained two transcriptionally distinct insulin-expressing tumour cell populations (INS+ and INS+FOS low ), characterised here for the first time. These two cancer cell populations significantly differed by ~ 8,000 differentially expressed genes (DEGs), particularly tumour suppressor genes (e.g. TP53, EGR1) and cancer-related pathways (e.g., MAPK, p53). In contrast, COX7A2L was one of a few genes ubiquitously expressed and significantly upregulated (> 20-fold) in both insulin-expressing tumour populations compared to other captured populations. Both populations were also characterised by expression of chromogranin/secretogranin neuroendocrine tumour marker genes (e.g. CHGA, SCGN). There were far fewer gene expression differences observed between insulin-expressing tumour cells from the two patients (~ 600 DEGs) than between the two cancer cell populations within each patient. These DEGs included CLTRN, TMSB4X, CSRP2, LGALS2, and C15orf48. Unexpectedly for a tumour of endocrine origin, the metastasis in Patient 2 exhibited > 20-70 fold upregulation of exocrine pancreatic genes including CLPS, PRSS2, PRSS and CTRC. Immune cell analyses identified distinct infiltrating immune populations, including memory T cells and macrophages and revealed likely tumour-immune interactions, including the CD40-CD40L interaction. This study provides the first single-cell RNA sequencing (scRNA-seq) analysis of naturally occurring insulinoma in any species, revealing tumour cell heterogeneity, novel immune microenvironment features, and potential therapeutic targets. Despite its small scale, the findings highlight the utility of scRNA-seq in veterinary oncology and its translational potential for pancreatic neuroendocrine tumours across species.
SUPPLEMENTARY INFORMATION: The online version contains supplementary material available at 10.1186/s44356-025-00026-3.
PMID:40438247 | PMC:PMC12106163 | DOI:10.1186/s44356-025-00026-3
Breaking the cycle of parasitic diseases with edutainment: The intersection of entertainment and education
PLoS Negl Trop Dis. 2025 May 28;19(5):e0013072. doi: 10.1371/journal.pntd.0013072. eCollection 2025 May.
ABSTRACT
Parasitic diseases represent a substantial public health challenge worldwide. Traditional educational strategies have often fallen short in driving sustained behavioral shifts that are nonetheless essential for reducing the burden of these diseases. Edutainment, a blend of education and entertainment, is the synthesis of pedagogical content with recreational frameworks, leveraging narrative and visual appeal to elevate the learning experience through enriched experiences, aligning with the principles of "warm cognition". Human cognitive processes, including attention, learning and memory, are influenced by emotions. As a result, emotional experiences are remembered vividly and accurately, with great resilience over time. Several edutainment approaches have been successfully utilized to inspire positive behavioral changes against soil-transmitted helminths (STHs), schistosomiasis, echinococcosis, and other diseases. This scoping review delves into several documented approaches with sustainable positive post-intervention outcomes. Approaches such as animated cartoons, gamification, songs, videos, and music, mobile health applications, hands-on experience, posters, comics and educational booklets, puppet shows, toy animals, cardboard and plastic-coated drawings, drawing activities and competitions, group discussions, illustrated booklets and questionnaires have yielded statistically significant improvements in participant's knowledge related to parasitic diseases (up to 60% increase in knowledge scores), alongside notable reductions in risks of parasite transmission and infection prevalence. These improvements highlight the potential of edutainment to enhance community awareness, promote long-term behavioral changes, and ultimately contribute to reducing spread of disease. Moreover, artificial intelligence (AI) can be integrated into edutainment approaches to meet the growing demand for personalized and effective learning methods. We argue that such AI-driven edutainment can underpin sustainable progress in the control of parasitic diseases.
PMID:40435280 | DOI:10.1371/journal.pntd.0013072
Epithelial damage and ageing: the perfect storm
Thorax. 2025 May 27:thorax-2024-222060. doi: 10.1136/thorax-2024-222060. Online ahead of print.
ABSTRACT
BACKGROUND: Idiopathic pulmonary fibrosis (IPF) is a progressive disease of lung parenchymal scarring that is triggered by repeated microinjury to a vulnerable alveolar epithelium. It is increasingly recognised that cellular ageing, whether physiological or accelerated due to telomere dysfunction, renders the epithelium less able to cope with injury and triggers changes in epithelial behaviour that ultimately lead to the development of disease.
AIMS: This review aims to highlight how, with increasing age, the alveolar epithelium becomes vulnerable to exogenous insults. We discuss the downstream consequences of alveolar epithelial dysfunction on epithelial phenotype, alveolar repair and pro-pathogenic interactions with other alveolar niche-resident cell types which drive IPF pathogenesis.
NARRATIVE: We highlight how a wide array of cellular mechanisms that maintain cellular homeostasis become dysfunctional with ageing. Waning replicative capacity, genomic stability, mitochondrial function, proteostasis and metabolic function all contribute to a phenotype of vulnerability to 'second hits'. We discuss how in IPF the alveolar epithelium becomes dysfunctional, highlighting changes in repair capacity and fundamental cellular phenotype and how interactions between abnormal epithelium and other alveolar niche-resident cell types perpetuate disease.
CONCLUSIONS: The ageing epithelium is a vulnerable epithelium which, with the cumulative effects of environmental exposures, fundamentally changes its behaviour towards stalled differentiation, failed repair and profibrotic signalling. Further dissection of aberrant epithelial behaviour, and its impact on other alveolar cell types, will allow identification of novel therapeutic targets aimed at earlier pathogenic events.
PMID:40425299 | DOI:10.1136/thorax-2024-222060
Clinical Pathology Administrator
Are you passionate about administration and supporting a Clinical Pathology team within a world-class research and education institution?
We have an exciting opportunity for a commercially minded Clinical Pathology Administrator to join our Clinical Pathology Laboratory at the Queen's Veterinary School Hospital. This is a full-time position working 36.5 hours a week, Monday to Friday.
The Queen's Veterinary School Hospital is a teaching and referral hospital with a reputation both nationally and internationally as a centre for excellence in many areas of veterinary medicine. The Clinical Pathology laboratory provides a range of services which are critical to the timely diagnosis of conditions of patients presenting in the clinics as well as being involved in a number of research projects. As a Clinical Pathology Administrator, you will manage the end-to-end process of sample handling and reporting, playing a key role in supporting the team and maintaining efficient workflows within the Clinical Pathology department. Your work ensures timely processing and accurate tracking, vital for delivering fast and reliable test results to clients.
This role is suitable for someone with qualifications comparable to an HND/HNC, Level 4/5 vocational qualifications, or equivalent practical experience. The ideal candidate will possess both factual and theoretical knowledge in administration and demonstrate a strong commercial mindset. Key responsibilities include evaluating processes, assessing pricing structures, and establishing efficient administrative methods. You would work with dedicated laboratory and practice management software, as well as routine word processing and spreadsheets.
Why Join us?
- Be a part of a prestigious institution renowned for its research and academic excellence
- Work in a collaborative and supportive environment
- 36 days annual leave per year including bank holidays
- A generous pension scheme
- Travel benefits, retail discounts at over 2,000 stores and much more via our benefits platform (Cambens)
- Contribute to the success of a department dedicated to delivering excellence in veterinary education and biomedical research
Informal enquiries can be made to Cassia Hare via email: chzh2@cam.ac.uk. If you have any queries regarding the application process, please contact Clinical HR via email: qvsh.hr@vet.cam.ac.uk.
Please quote reference PP46064 on your application and in any correspondence about this vacancy. Further particulars for the role and information about the Department can be found on www.vet.cam.ac.uk.
Click the 'Apply' button below to register an account with our recruitment system (if you have not already) and apply online.
Closing date: Sunday, 8 June 2025
We reserve the right to close this vacancy early if we receive sufficient applications or extend it if we do not receive a sufficient number of applications. Therefore, if you are interested, please submit your application as early as possible.
The University actively supports equality, diversity and inclusion and encourages applications from all sections of society.
The University has a responsibility to ensure that all employees are eligible to live and work in the UK.
Opportunities and challenges for monitoring terrestrial biodiversity in the robotics age
Nat Ecol Evol. 2025 May 22. doi: 10.1038/s41559-025-02704-9. Online ahead of print.
ABSTRACT
With biodiversity loss escalating globally, a step change is needed in our capacity to accurately monitor species populations across ecosystems. Robotic and autonomous systems (RAS) offer technological solutions that may substantially advance terrestrial biodiversity monitoring, but this potential is yet to be considered systematically. We used a modified Delphi technique to synthesize knowledge from 98 biodiversity experts and 31 RAS experts, who identified the major methodological barriers that currently hinder monitoring, and explored the opportunities and challenges that RAS offer in overcoming these barriers. Biodiversity experts identified four barrier categories: site access, species and individual identification, data handling and storage, and power and network availability. Robotics experts highlighted technologies that could overcome these barriers and identified the developments needed to facilitate RAS-based autonomous biodiversity monitoring. Some existing RAS could be optimized relatively easily to survey species but would require development to be suitable for monitoring of more 'difficult' taxa and robust enough to work under uncontrolled conditions within ecosystems. Other nascent technologies (for instance, new sensors and biodegradable robots) need accelerated research. Overall, it was felt that RAS could lead to major progress in monitoring of terrestrial biodiversity by supplementing rather than supplanting existing methods. Transdisciplinarity needs to be fostered between biodiversity and RAS experts so that future ideas and technologies can be codeveloped effectively.
PMID:40404926 | DOI:10.1038/s41559-025-02704-9
Cambridge researchers named as 2025 Academy of Medical Sciences Fellows
The new Fellows have been recognised for their remarkable contributions to advancing medical science, groundbreaking research discoveries and translating developments into benefits for patients and the wider public. Their work exemplifies the Academy’s mission to create an open and progressive research sector that improves health for everyone.
They join an esteemed Fellowship of 1,450 researchers who are at the heart of the Academy’s work, which includes nurturing the next generation of scientists and shaping research and health policy in the UK and worldwide.
One of Cambridge’s new Fellows, Professor Sam Behjati, is a former recipient of the Academy’s prestigious Foulkes Foundation medal, which recognises rising stars within biomedical research. Sam is Clinical Professor of Paediatric Oncology at the University and an Honorary Consultant Paediatric Oncologist at Addenbrooke’s Hospital, as well as Group Leader at the Wellcome Sanger Institute. His research is rooted in cancer genomics, phylogenetics, and single cell transcriptomics and spans a wide range of diseases and biological problems. More recently, his work has focused on the origin of cancers, in particular of childhood cancer. In addition, he explores how to use genomic data to improve the treatment of children. Sam is a Fellow at Corpus Christi College, Cambridge.
Also elected to the Academy of Medical Sciences Fellowship are:
Professor Clare Bryant, Departments of Medicine and Veterinary Medicine
Clare Bryant is Professor of Innate Immunity. She studies innate immune cell signalling during bacterial infection to answer fundamental questions about host-pathogen interactions and to search for new drugs to modify them. She also applies these approaches to study inflammatory signalling in chronic diseases of humans and animals. Clare has extensive collaborations with many pharmaceutical companies, is on the scientific advisory board of several biotech companies, and helped found the natural product company Polypharmakos. Clare is a Fellow of Queens’ College, Cambridge.
Professor Frank Reimann, Institute of Metabolic Science-Metabolic Research Laboratories
Frank Reimann is Professor of Endocrine Signaling. The main focus of his group, run in close partnership with Fiona Gribble, is the enteroendocrine system within the gut, which helps regulate digestion, metabolism, and how full we feel. Their work has included the use of animal models and human cellular models to understand how cells function. One of these cells, glucagon-like peptide-1 (GLP-1) is the target of therapies now widely used in the treatment of diabetes mellitus and obesity. How cells shape feeding behaviour has become a major focus of the lab in recent years.
Professor Mina Ryten, UK Dementia Research Institute
Mina Ryten is a clinical geneticist and neuroscientist, and Director of the UK Dementia Research Institute at Cambridge since January 2024. She also holds the Van Geest Professorship and leads a lab focused on understanding molecular mechanisms driving neurodegeneration. Mina’s research looks at how genetic variation influences neurological diseases, particularly Lewy body disorders. Her work has advanced the use of single cell and long-read RNA sequencing to map disease pathways and identify potential targets for new treatments. Her expertise in clinical care and functional genomics has enabled her to bridge the gap between patient experience and scientific discovery.
Professor Andrew Morris CBE FRSE PMedSci, President of the Academy of Medical Sciences, said: “The breadth of disciplines represented in this year’s cohort – from mental health and infectious disease to cancer biology and respiratory medicine – reflects the rich diversity of medical science today. Their election comes at a crucial time when scientific excellence and collaboration across disciplines are essential for addressing global health challenges both now and in the future. We look forward to working with them to advance biomedical research and create an environment where the best science can flourish for the benefit of people everywhere.”
The new Fellows will be formally admitted to the Academy at a ceremony on Wednesday 9 July 2025.
Four Cambridge biomedical and health researchers are among those announced today as newly-elected Fellows of the Academy of Medical Sciences.
Big T Images for Academy of Medical SciencesAcademy of Medical Sciences plaque
The text in this work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. Images, including our videos, are Copyright ©University of Cambridge and licensors/contributors as identified. All rights reserved. We make our image and video content available in a number of ways – on our main website under its Terms and conditions, and on a range of channels including social media that permit your use and sharing of our content under their respective Terms.
Visual preferences for communicating modelling: a global analysis of COVID-19 policy and decision makers
Infect Dis Model. 2025 Apr 23;10(3):924-934. doi: 10.1016/j.idm.2025.04.005. eCollection 2025 Sep.
ABSTRACT
Effective communication of modelling results to policy and decision makers has been a longstanding challenge in times of crises. This communication takes many forms - visualisations, reports, presentations - and requires careful consideration to ensure accurate maintenance of the key scientific messages. Science-to-policy communication is further exacerbated when presenting fundamentally uncertain forms of science such as infectious disease modelling and other types of modelled evidence, something which has been understudied. Here we assess the communication and visualisation of infectious disease modelling results to national COVID-19 policy and decision makers in 13 different countries. We present a synthesis of recommendations on what aspects of visuals, graphs, and plots policymakers found to be most helpful in their COVID-19 response work. This work serves as a first evidence base for developing guidelines on the communication and translation of infectious disease modelling into policy.
PMID:40390802 | PMC:PMC12088752 | DOI:10.1016/j.idm.2025.04.005
Competition between transmission lineages mediated by human mobility shapes seasonal influenza epidemics in the US
Nat Commun. 2025 May 17;16(1):4605. doi: 10.1038/s41467-025-59757-4.
ABSTRACT
Due to its climatic variability, complex mobility networks and geographic expanse, the United States represents a compelling setting to explore the transmission processes that lead to heterogeneous yearly seasonal influenza epidemics. By analyzing genomic and epidemiological data collected in the US from 2014 to 2023, we show that epidemics consisted of multiple co-circulating transmission lineages that could emerge from all regions and often rapidly expanded. Lineage spread was characterized by strong spatiotemporal hierarchies and lineage size correlated with timing of establishment in the US. Mechanistic epidemic simulations, supported by phylogeographic analyses, suggest that competition between lineages on a network of human mobility consistent with commuting flows drove lineage dynamics. Our results suggest that the processes that disseminate viruses nationwide are highly structured, but variability in the short-term processes that determine the locations, timing, and explosiveness of initial epidemic sparks limits predictability of regional and national epidemics.
PMID:40382319 | DOI:10.1038/s41467-025-59757-4
Development of a Novel Epilepsy and Dyskinesia Survey for Large-Scale Characterization of Seizure Semiology in Dogs
J Vet Intern Med. 2025 May-Jun;39(3):e70077. doi: 10.1111/jvim.70077.
ABSTRACT
BACKGROUND: Diagnosing epilepsy and dyskinesia in dogs relies on seizure semiology, laboratory workup, brain imaging, and electroencephalography. Variability in existing epilepsy surveys complicates comparison and impedes epidemiologic and genetic research.
OBJECTIVE: To characterize the semiology of epileptic seizures and dyskinesia episodes using a novel, owner-completed, multi-language online questionnaire.
ANIMALS: A cohort of 606 dogs from 96 breeds with paroxysmal events, perceived by their owners as epilepsy or dyskinesia.
MATERIALS AND METHODS: A comprehensive epilepsy and dyskinesia questionnaire featuring pragmatic seizure categories and video upload was developed in German, Finnish, and English. The reliability of the questionnaire was assessed, and the study cohort analyzed.
RESULTS: The questionnaire demonstrated strong internal consistency and interrater agreement. Owners correctly classified paroxysmal events in 90.1% of cases (95% CI 88.18-92.11). Video footage was submitted from 23.8% (143/606) and supported the seizure type in the questionnaire in 96.5%. The age of onset ranged from 6 months to 6 years in 80.2% (median 2 years; IQR 1-5 years). Generalized (epileptic) convulsive seizures occurred in 58.6% of dogs, non-generalized paroxysmal motor events without convulsions in 58.1%, sudden falls without movement in 6.1%, episodes of impaired awareness in 15.8%, and other unclassified events in 7.1%. Multiple seizure types were reported in 25.2% of the dogs. Labrador Retrievers exhibited a higher prevalence of non-generalized motor events compared to Border Collies, Siberian Huskies, and other breeds (p < 0.001).
CONCLUSIONS: The questionnaire reliably characterizes epileptic seizures and dyskinesia episodes in dogs, making it a valuable tool for large-scale epidemiological and genetic studies.
PMID:40375574 | DOI:10.1111/jvim.70077
Static respiratory compliance in anaesthetised and intubated brachycephalic dogs with and without brachycephalic obstructive airway syndrome
Vet J. 2025 May 13:106372. doi: 10.1016/j.tvjl.2025.106372. Online ahead of print.
ABSTRACT
The impact of brachycephalic obstructive airway syndrome in dogs (BOAS) on respiratory mechanics is unclear and may affect the choice of ventilation strategies during anaesthesia. This prospective study included 56 client-owned brachycephalic dogs, allocated to be BOAS (n = 26) or non-BOAS dogs (n = 30) based on functional grading. All dogs were anaesthetised using a standardised anaesthetic protocol. Pressure-controlled ventilation was initiated around 30minutes post-induction, maintaining peak inspiratory pressure at 7-12cm H2O. Static respiratory compliance (Cstat) was recorded at predetermined time points in sternal, right and left lateral recumbency. Thorax dimensions were assessed with a tape measure. Body surface area (BSA) was calculated and the ratio Cstat/BSA used as the main outcome variable. Comparison of means/medians, analysis of proportions, the Spearman correlation coefficient and both logistic and linear regression were used for data analysis. P < 0.05 was considered statistically significant. Non-BOAS dogs showed significantly higher Cstat/BSA compared to BOAS dogs in sternal (41.6 (31.1-51.8) vs. 32.9 (24.4 - 39.2), respectively, P = 0.028), right lateral (36.2 (25.7 - 46.4) vs. 27.0 (22.7 - 35.6); P = 0.026) and left lateral (33.6 (22.6 - 45.5) vs. 24.6 (18.4 - 32.2); P = 0.020) recumbencies. For all dogs, the Cstat/BSA ratio was higher in sternal compared to lateral recumbencies. BOAS dogs had a significantly shorter distance between thoracic inlet and last rib compared to non-BOAS dogs (20 ± 4 vs. 23 ± 6cm, respectively; P = 0.043). Reduced respiratory compliance in BOAS-affected dogs should be considered during mechanical ventilation.
PMID:40374099 | DOI:10.1016/j.tvjl.2025.106372